

Cholinergic modulation of angiogenesis: role of the 7 nicotinic acetylcholine receptor
- PMID: 19623583
- PMCID: PMC3140170
- DOI: 10.1002/jcb.22270
Cholinergic modulation of angiogenesis: role of the 7 nicotinic acetylcholine receptor
Abstract
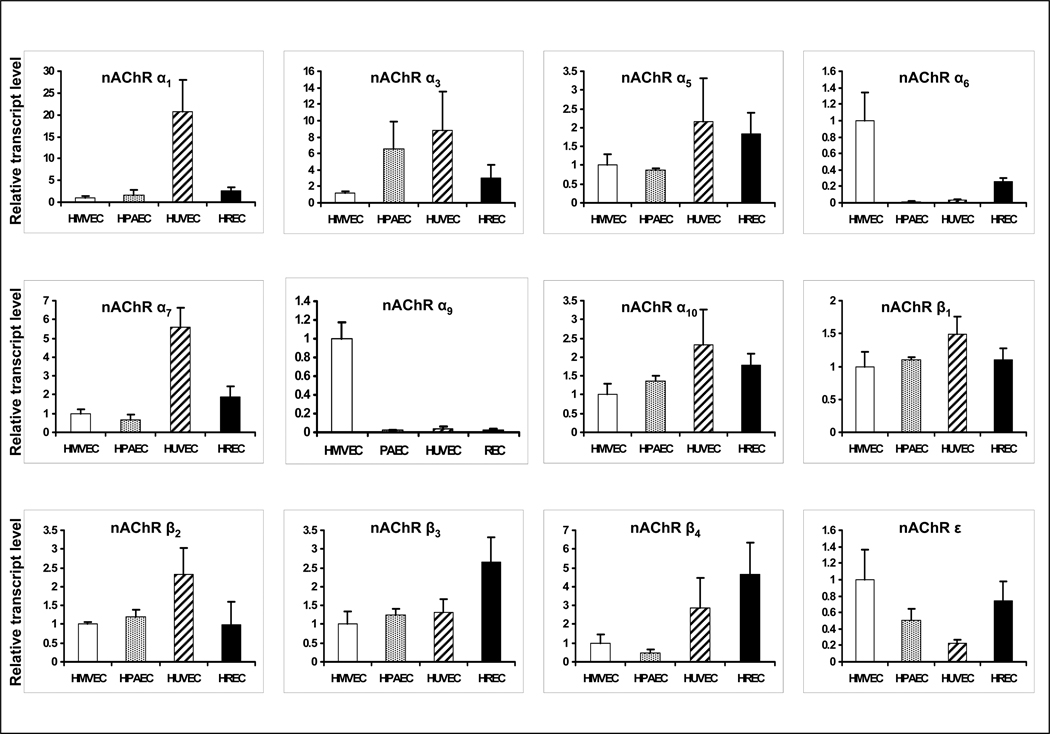
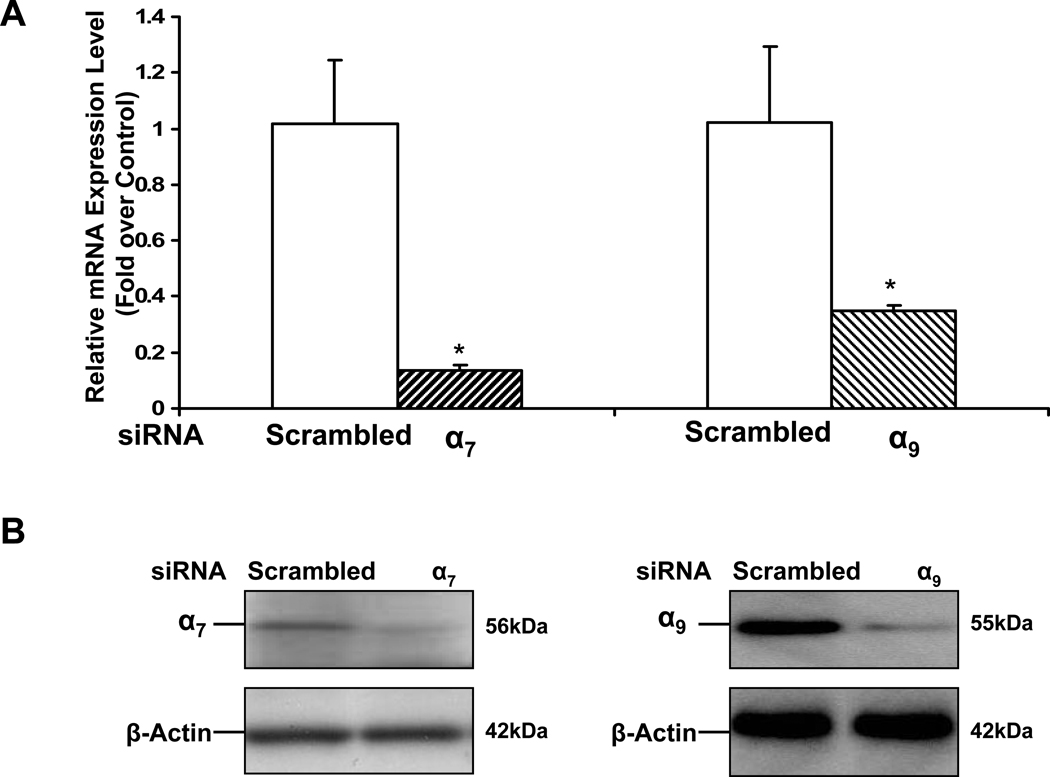
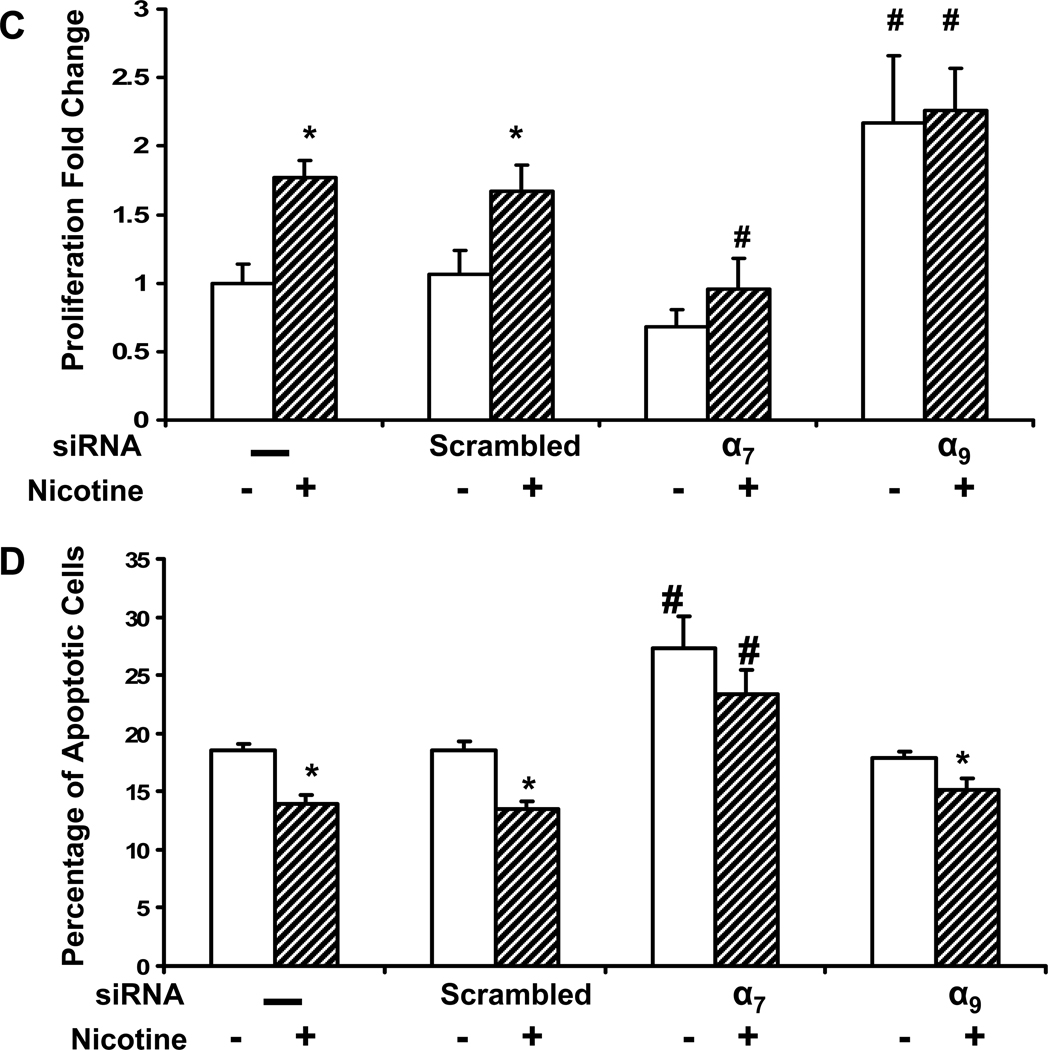
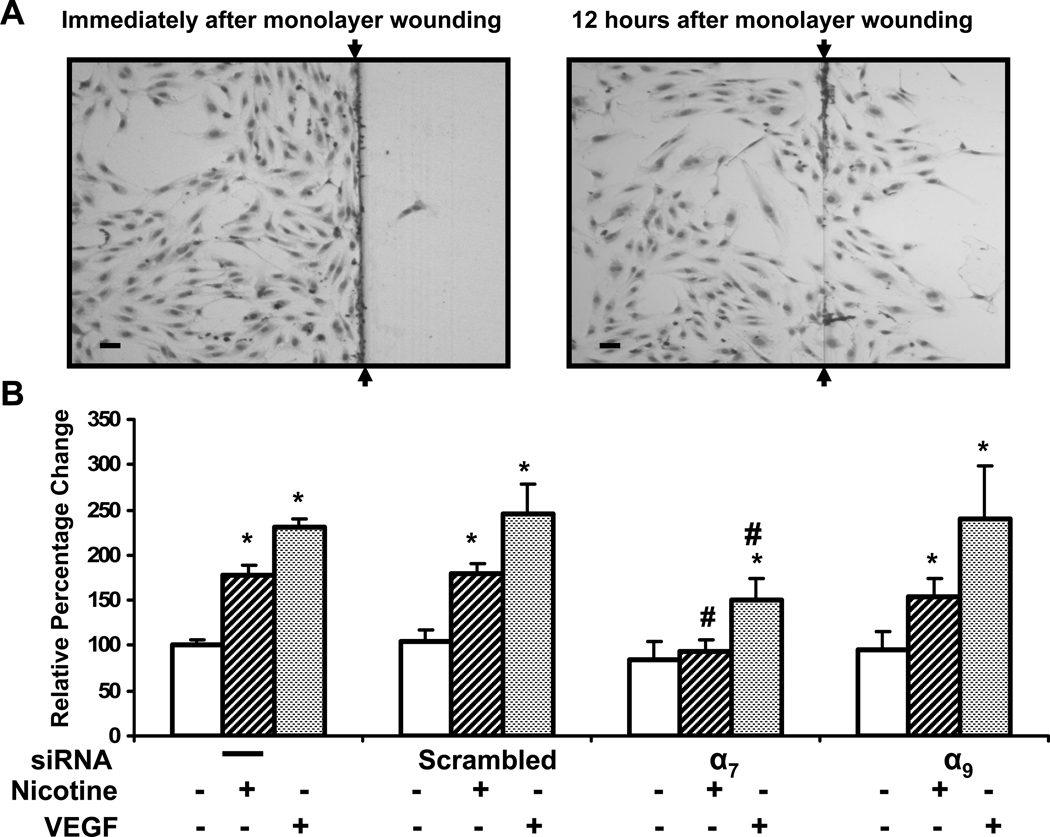
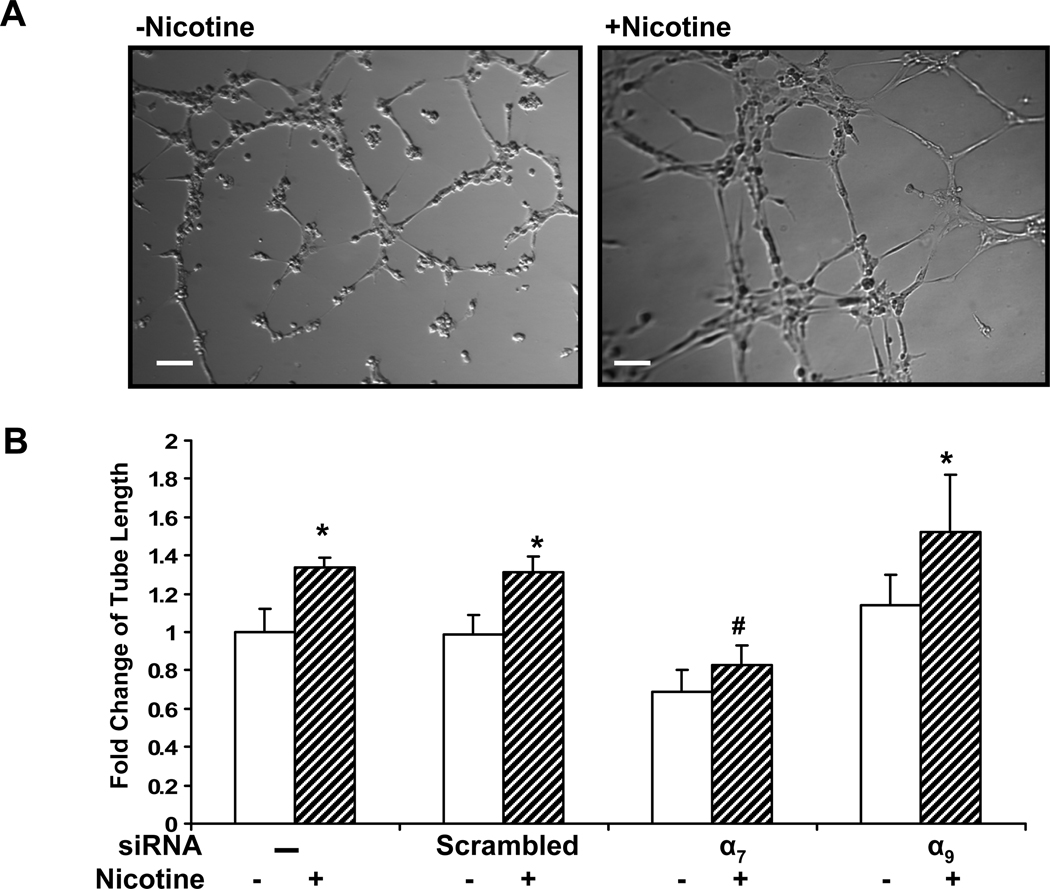
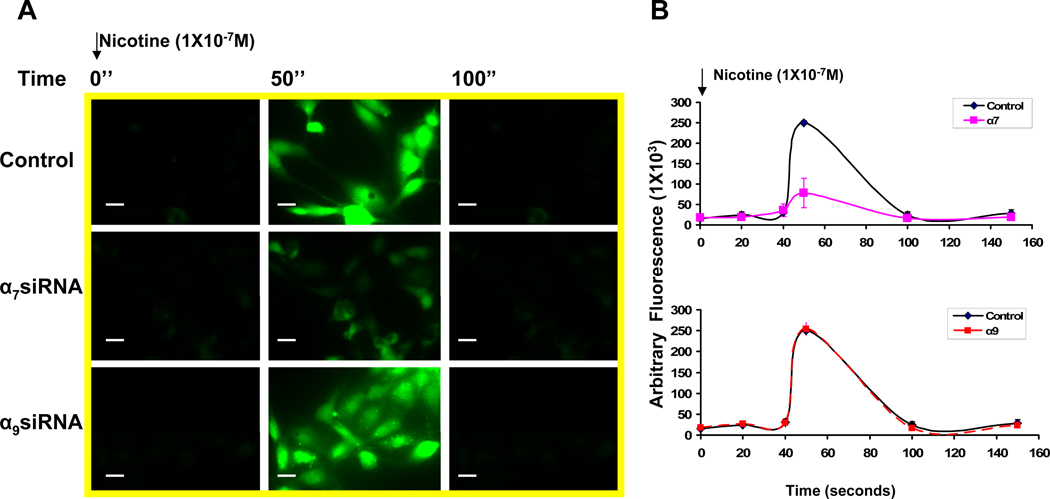
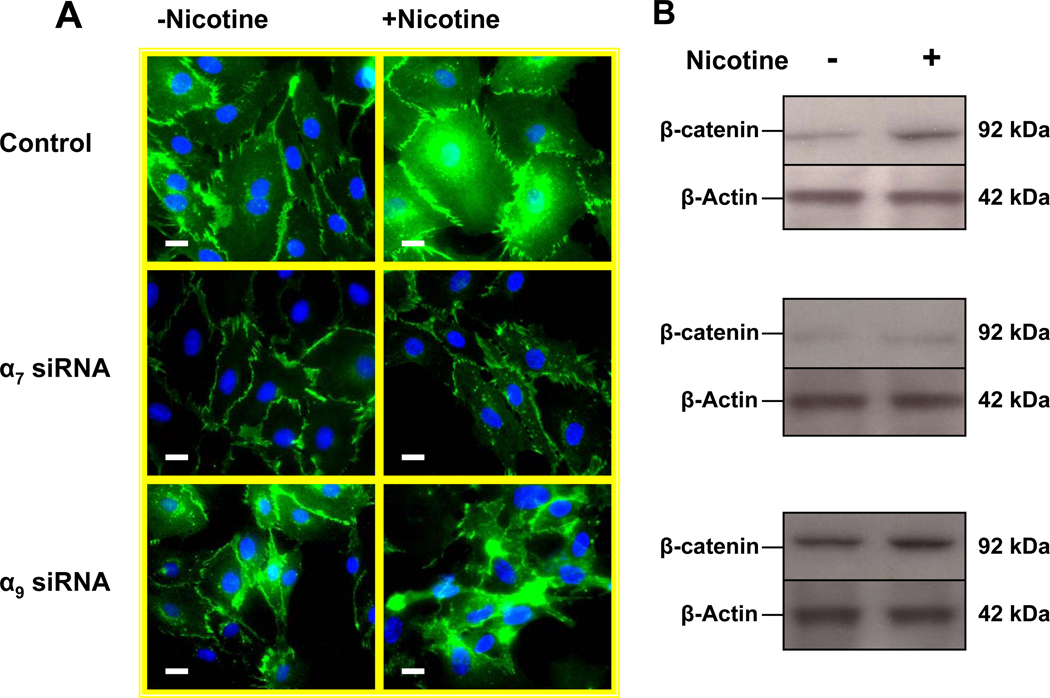
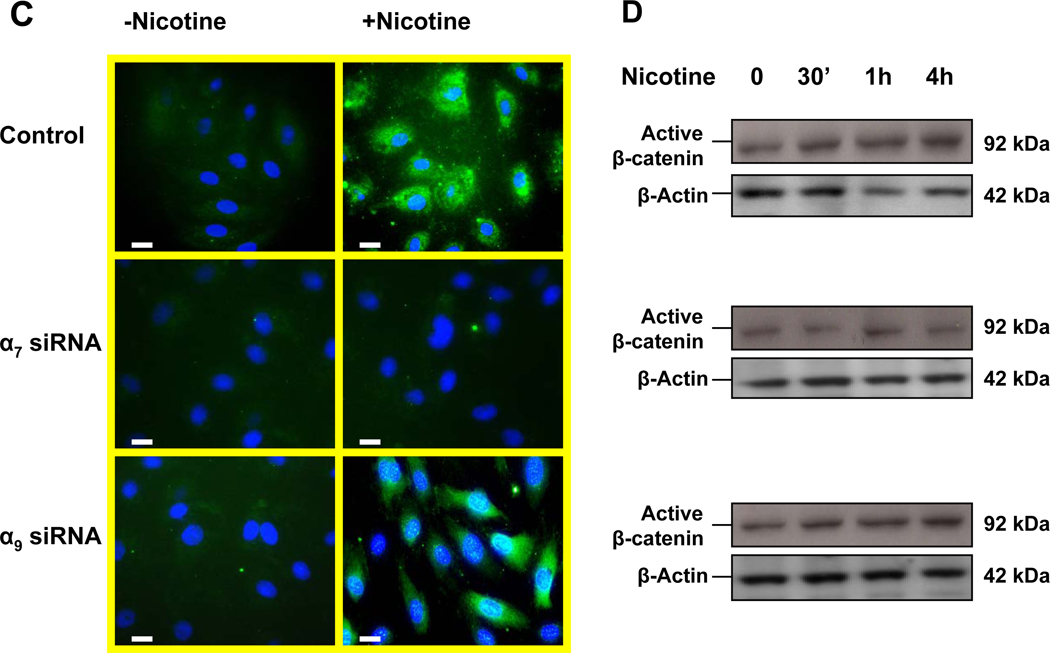
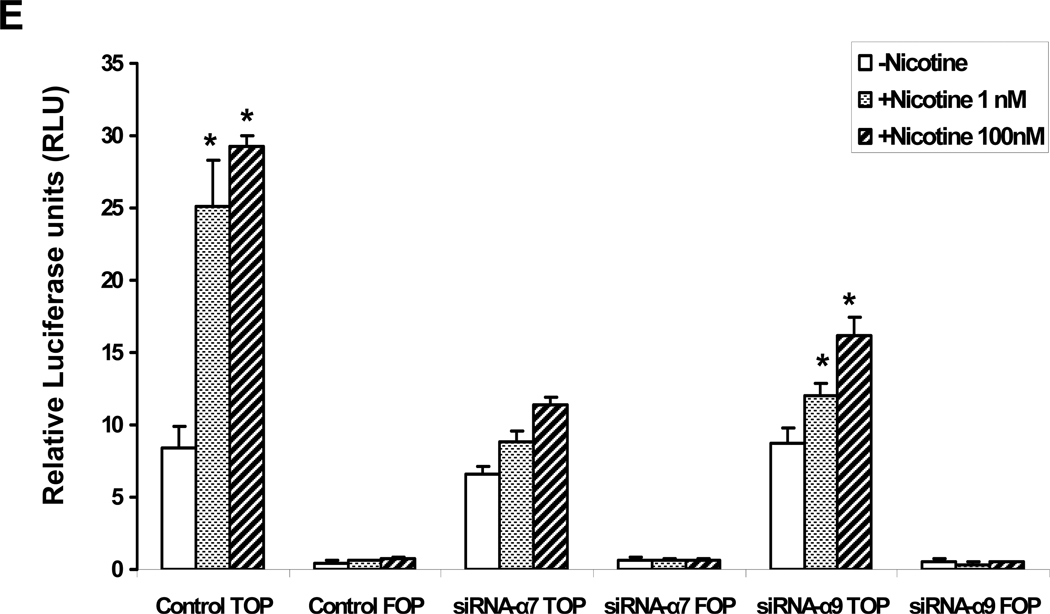
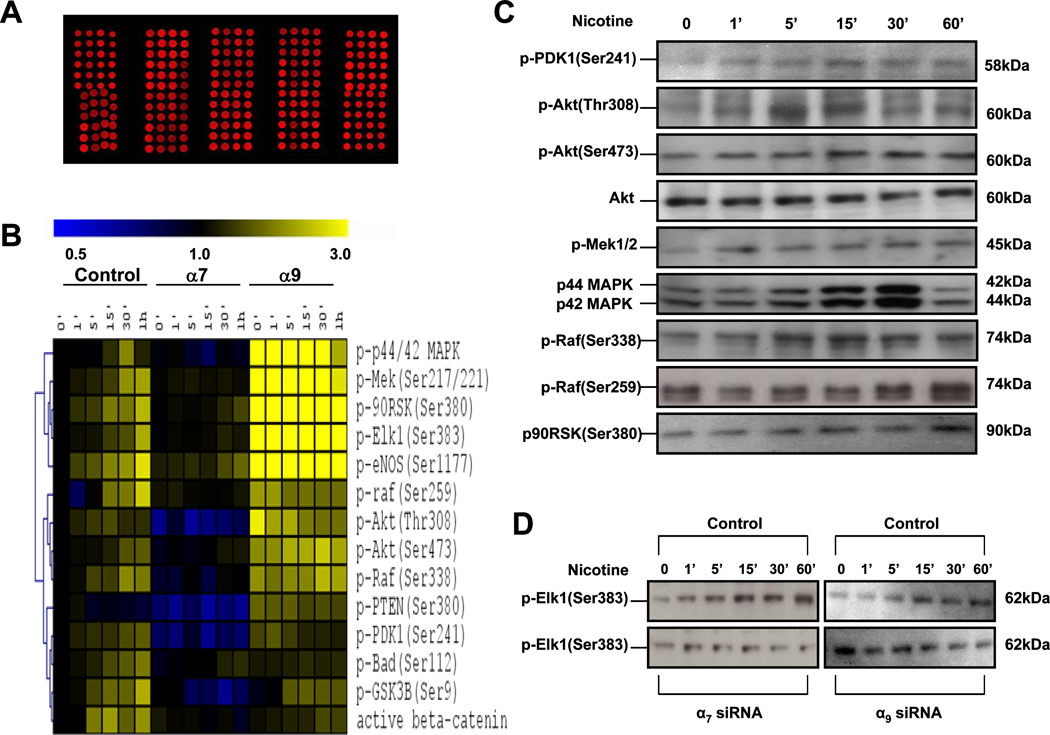
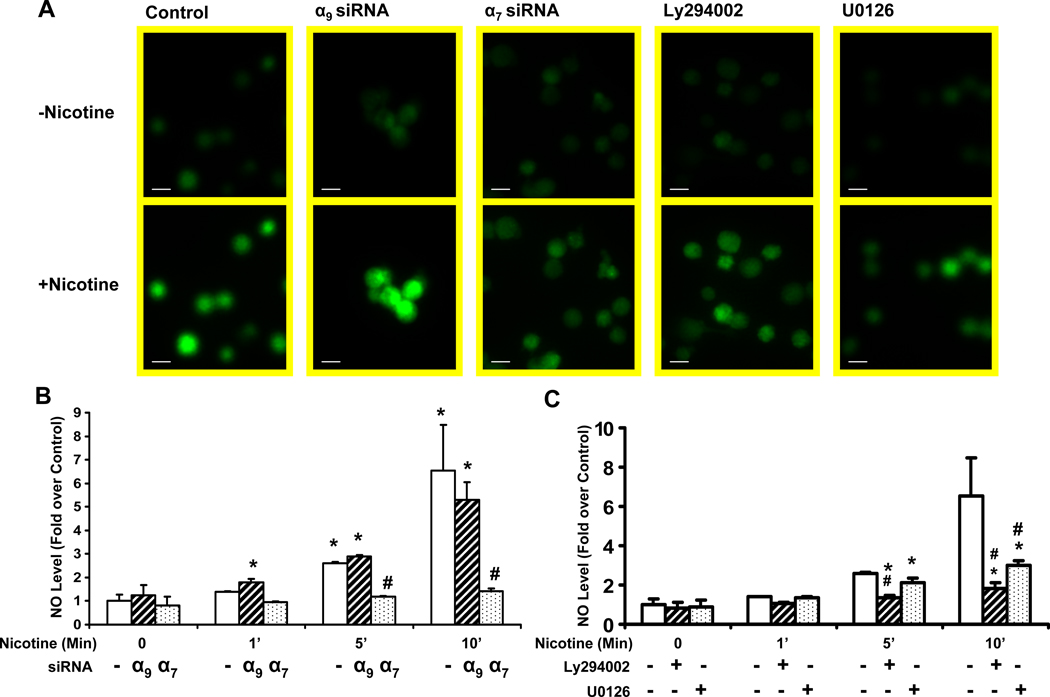
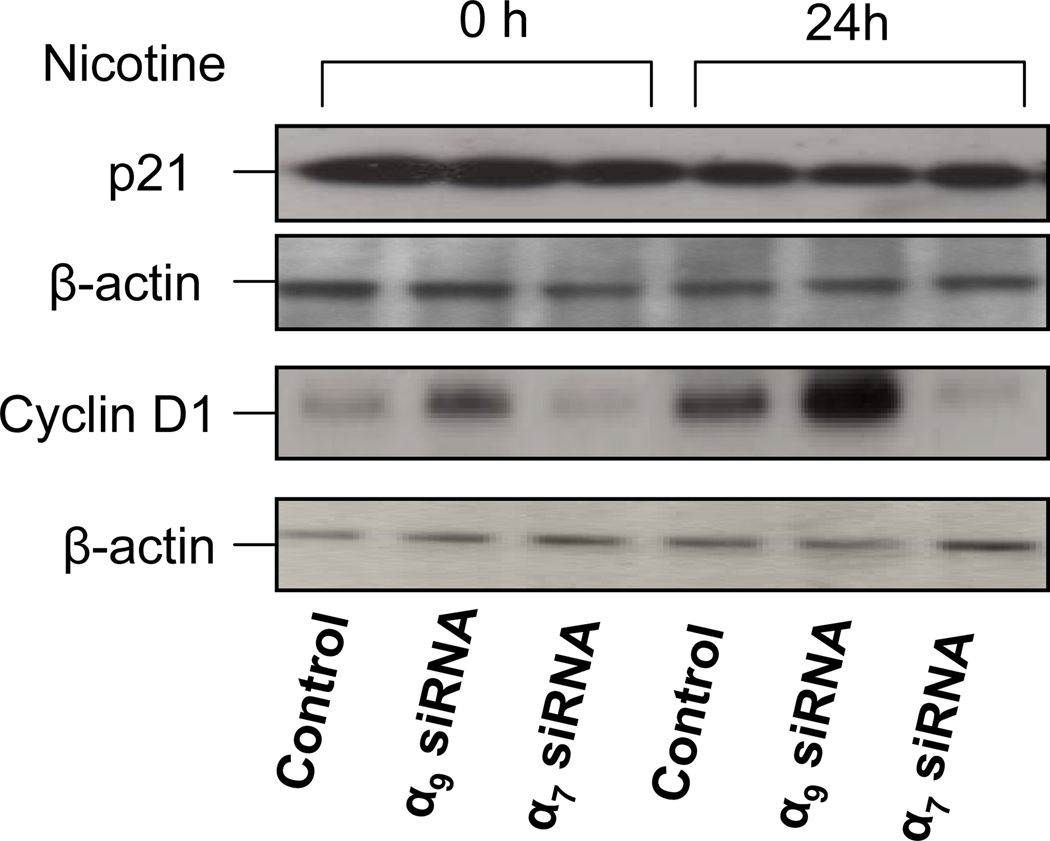
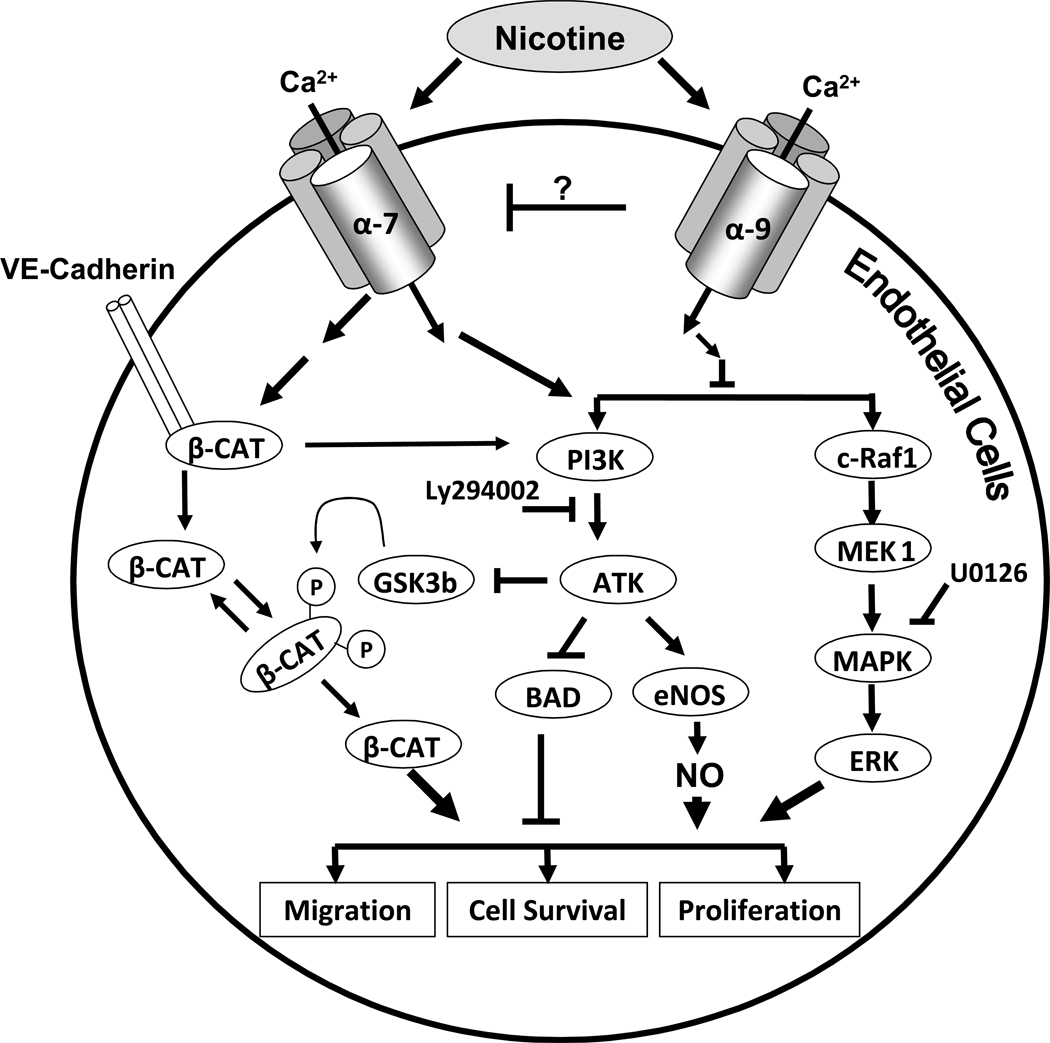
Pathological angiogenesis contributes to tobacco-related diseases such as malignancy, atherosclerosis and age-related macular degeneration. Nicotine acts on endothelial nicotinic acetylcholine receptors (nAChRs) to activate endothelial cells and to augment pathological angiogenesis. In the current study, we studied nAChR subunits involved in these actions. We detected mRNA for all mammalian nAChR subunits except alpha(2), alpha(4), gamma, and delta in four different types of ECs. Using siRNA methodology, we found that the alpha(7) nAChR plays a dominant role in nicotine-induced cell signaling (assessed by intracellular calcium and NO imaging, and studies of protein expression and phosphorylation), as well as nicotine-activated EC functions (proliferation, survival, migration, and tube formation). The alpha(9) and alpha(7) nAChRs have opposing effects on nicotine-induced cell proliferation and survival. Our studies reveal a critical role for the alpha(7) nAChR in mediating the effects of nicotine on the endothelium. Other subunits play a modulatory role. These findings may have therapeutic implications for diseases characterized by pathological angiogenesis.
(c) 2009 Wiley-Liss, Inc.
Figures
References
-
- Barbieri Ruggiero, Tremoli, Weksler Suppressing PTEN Activity by Tobacco Smoke Plus Interleukin-1 β Modulates Dissociation of VE-Cadherin/β-Catenin Complexes in Endothelium. Arteriosclerosis, thrombosis, and vascular biology. 2008 - PubMed
-
- Barbieri SS, Weksler BB. Tobacco smoke cooperates with interleukin-1beta to alter beta-catenin trafficking in vascular endothelium resulting in increased permeability and induction of cyclooxygenase-2 expression in vitro and in vivo. The FASEB journal. 2007;21:1831–1843. - PubMed
-
- Behrens J, von Kries JP, Kahl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. - PubMed
-
- Chan SM, Ermann J, Su L, Fathman CG, Utz PJ. Protein microarrays for multiplex analysis of signal transduction pathways. Nature medicine. 2004;10:1390–1396. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
