

doi: 10.1186/gb-2009-10-7-r79.
Epub 2009 Jul 22.
Methods for analyzing deep sequencing expression data: constructing the human and mouse promoterome with deepCAGE data
Affiliations
- PMID: 19624849
- PMCID: PMC2728533
- DOI: 10.1186/gb-2009-10-7-r79
Item in Clipboard
Methods for analyzing deep sequencing expression data: constructing the human and mouse promoterome with deepCAGE data
Genome Biol.
2009.
Abstract
With the advent of ultra high-throughput sequencing technologies, increasingly researchers are turning to deep sequencing for gene expression studies. Here we present a set of rigorous methods for normalization, quantification of noise, and co-expression analysis of deep sequencing data. Using these methods on 122 cap analysis of gene expression (CAGE) samples of transcription start sites, we construct genome-wide 'promoteromes' in human and mouse consisting of a three-tiered hierarchy of transcription start sites, transcription start clusters, and transcription start regions.
Figures
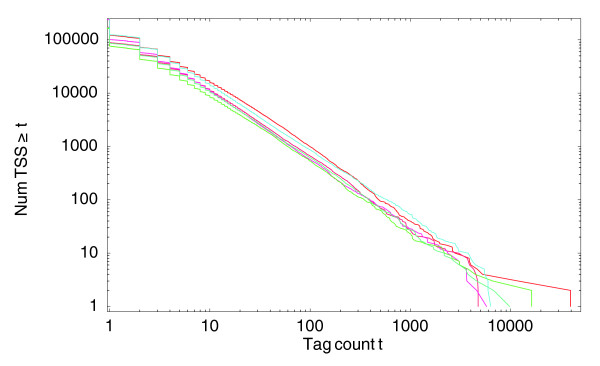
Reverse cumulative distributions for the number of different TSS positions that have at least a given number of tags mapping to them. Both axes are shown on a logarithmic scale. The three red curves correspond to the distributions of the three THP-1 cell control samples and the three blue curves to the three THP-1 samples after 24 hours of phorbol myristate acetate treatment. All other samples show very similar distributions (data not shown).
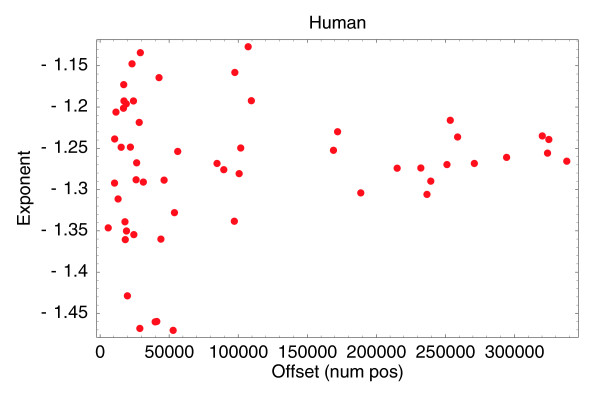
Fitted off-sets n0 (horizontal axis) and fitted exponents α (vertical axis) for the 56 human CAGE samples that have at least 100,000 tags.
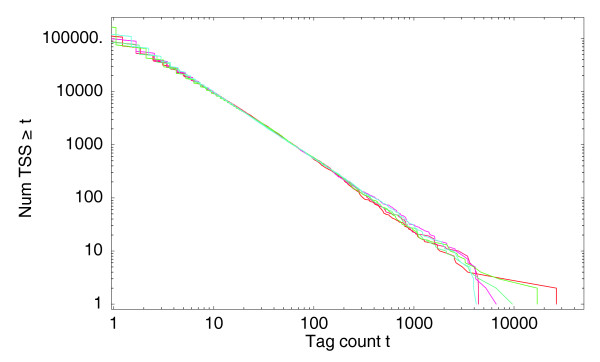
Normalized reverse cumulative distributions for the number of different TSS positions that have at least a given number of tags mapping to them. Both axes are shown on a logarithmic scale. The three red curves correspond to the distributions of the three THP-1 control samples and the three blue curves to the three THP-1 samples after 24 hours of PMA treatment.

CAGE replicate from THP-1 cells after 8 hours of lipopolysaccharide treatment. For each position with mapped tags, the logarithm of the number of tags per million (TPM) in the first replicate is shown on the horizontal axis, and the logarithm of the number of TPM in the second replicate on the vertical axis. Logarithms are natural logarithms.
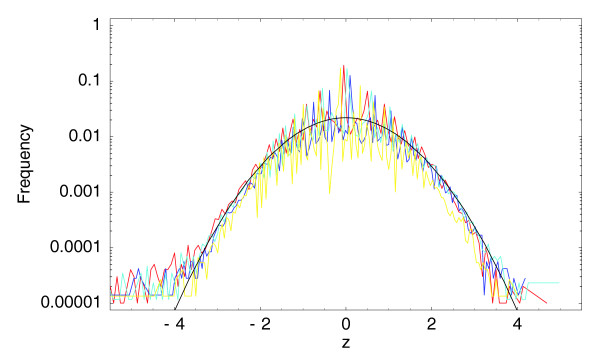
Observed histograms of z-statistics for the three 0/1 hour (in red, dark blue, and light blue) samples and for the technical replicate (in yellow) compared with the standard unit Gaussian (in black). The vertical axis is shown on a logarithmic scale.
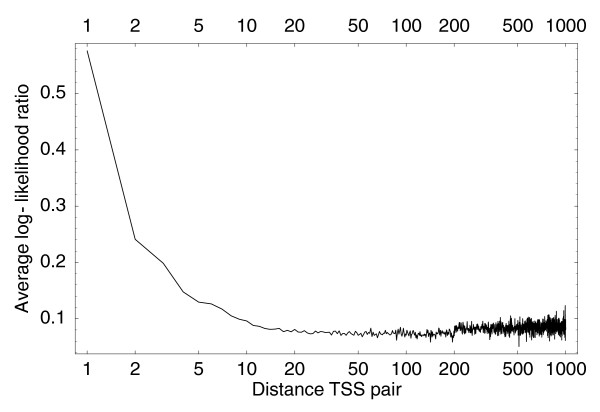
Average log-ratio L (Equation 13) for neighboring pairs of individual TSSs as a function of the distance between the TSSs. The horizontal axis is shown on a logarithmic scale.
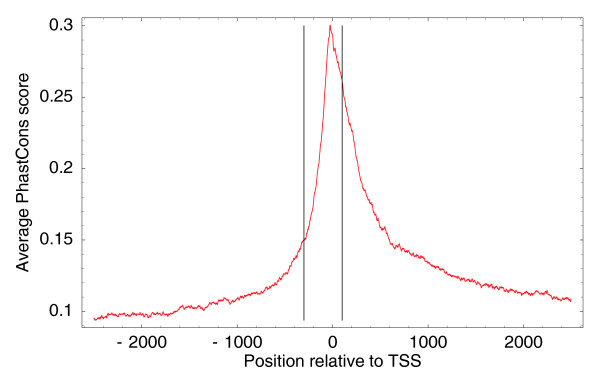
Average PhastCons (conservation) score relative to TSSs of genomic regions upstream and downstream of all human TSCs. The vertical lines show positions -300 and +100 with respect to TSSs.
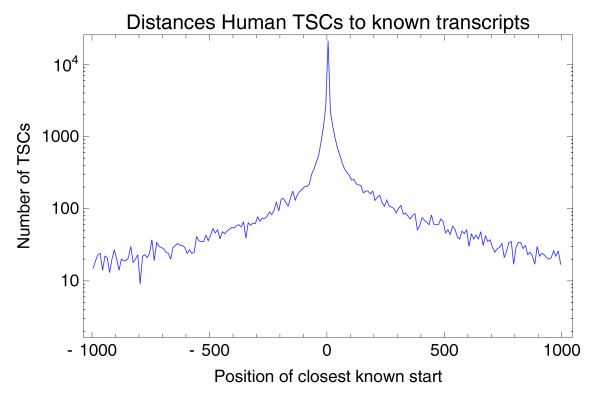
The number of TSCs as a function of their position relative to the nearest known mRNA start. Negative numbers mean the nearest known mRNA start is upstream of the TSC. The vertical axis is shown on a logarithmic scale. The figure shows only the 46,293 TSCs (62.3%) that have a known mRNA start within 1,000 bp.
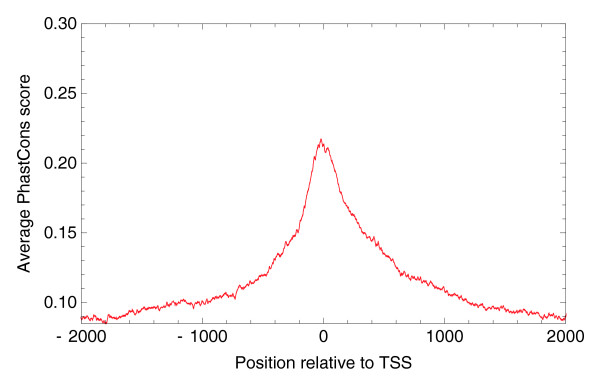
Average PhastCons (conservation) score relative to TSSs of genomic regions upstream and downstream of 'novel' human TSCs that are more than 5 kb away from the start of any known transcript.

Hierarchical structure of the human promoterome. (a) Distribution of the number of TSSs per co-expressed TSC. (b) Distribution of the number of TSCs per TSR. (c) Distribution of the number of TSSs per TSR. The vertical axis is shown on a logarithmic scale in all panels. The horizontal axis is shown on a logarithmic scale in (a, c).
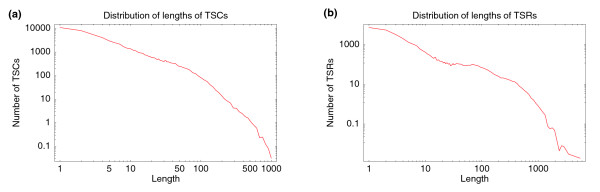
Length (base pairs along the genome) distribution of (a) TSCs and (b) TSRs. Both axes are shown on logarithmic scales in both panels.

Nearby TSCs with significantly differing expression profiles. (a) A 90-bp region on chromosome 3 containing 5 TSCs (colored segments) and the start of the annotated locus of the SENP5 gene (black segment). (b) Positions of the individual TSSs in the TSC and their total expression, colored according to the TSC to which each TSS belongs. (c) Expression across the 56 CAGE samples for the red and blue colored TSCs.
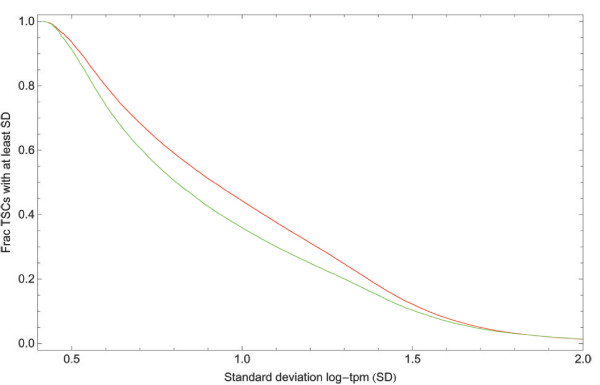
Reverse cumulative distributions of the standard deviation in expression across the 56 CAGE samples for the TSCs obtained with our clustering procedure (red) and the FANTOM3 single-linkage clustering procedure (green).

Two-dimensional histogram (shown as a heatmap) of the CG base content (horizontal axis) and CpG dinucleotide content (vertical axis) of all human TSRs. Both axes are shown on logarithmic scales.
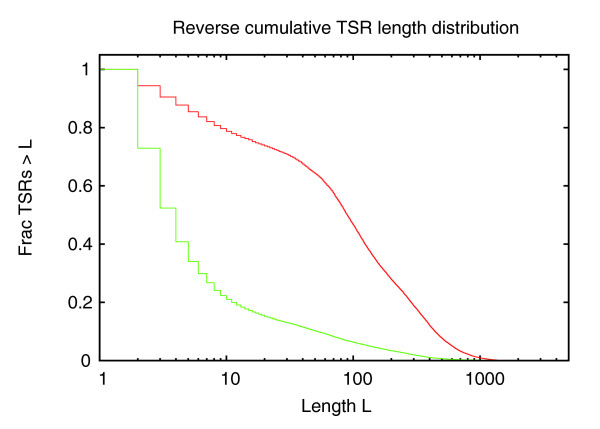
Reverse cumulative distribution of the lengths (base-pairs along the genome) of TSRs for high-CpG (red curve) and low-CpG (green curve) promoters. The horizontal axis is shown on a logarithmic scale.
References
-
- Maeda N, Nishiyori H, Nakamura M, Kawazu C, Murata M, Sano H, Hayashida K, Fukuda S, Tagami M, Hasegawa A, Murakami K, Schroder K, Hume KID, Hayashizaki Y, Carninci P, Suzuki H. Development of a DNA barcode tagging method for monitoring dynamic changes in gene expression by using an ultra high-throughput sequencer. Biotechniques. 2008;45:95–97. doi: 10.2144/000112814. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
