

Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge
- PMID: 19625450
- PMCID: PMC2743625
- DOI: 10.1091/mbc.e09-01-0041
Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge
Abstract
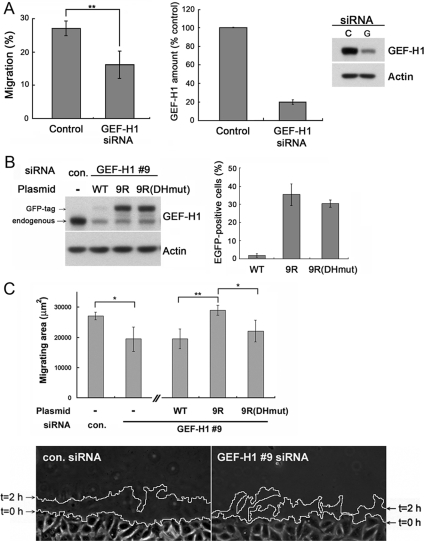
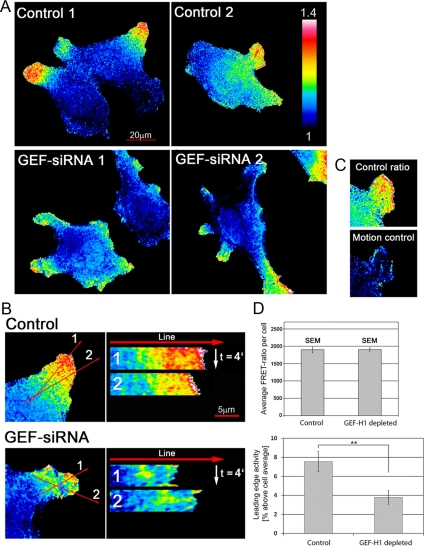
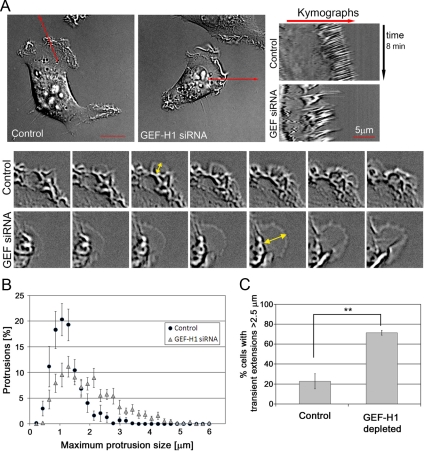
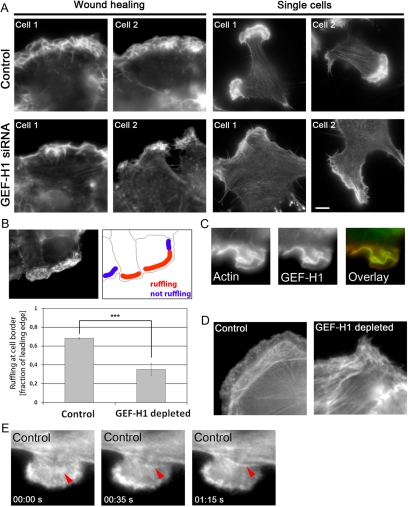
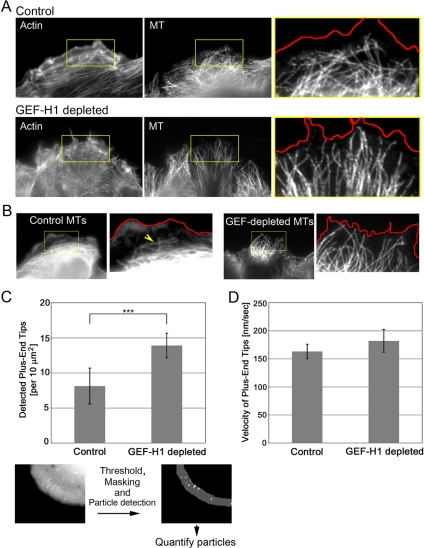
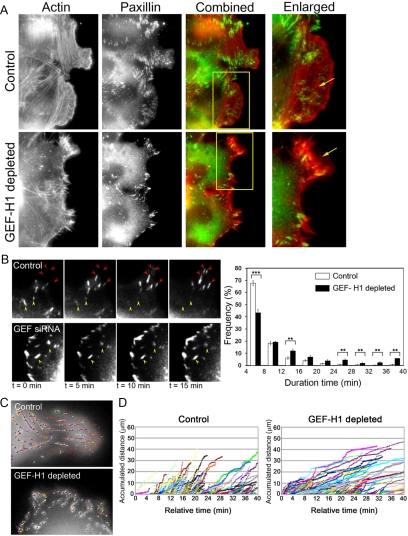
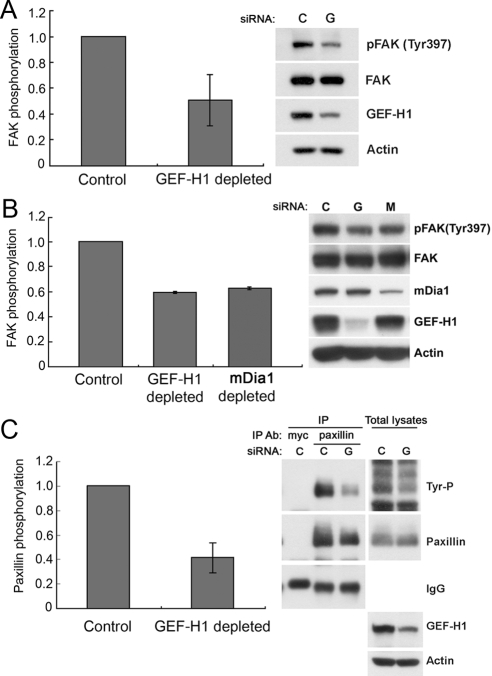
Cell migration involves the cooperative reorganization of the actin and microtubule cytoskeletons, as well as the turnover of cell-substrate adhesions, under the control of Rho family GTPases. RhoA is activated at the leading edge of motile cells by unknown mechanisms to control actin stress fiber assembly, contractility, and focal adhesion dynamics. The microtubule-associated guanine nucleotide exchange factor (GEF)-H1 activates RhoA when released from microtubules to initiate a RhoA/Rho kinase/myosin light chain signaling pathway that regulates cellular contractility. However, the contributions of activated GEF-H1 to coordination of cytoskeletal dynamics during cell migration are unknown. We show that small interfering RNA-induced GEF-H1 depletion leads to decreased HeLa cell directional migration due to the loss of the Rho exchange activity of GEF-H1. Analysis of RhoA activity by using a live cell biosensor revealed that GEF-H1 controls localized activation of RhoA at the leading edge. The loss of GEF-H1 is associated with altered leading edge actin dynamics, as well as increased focal adhesion lifetimes. Tyrosine phosphorylation of focal adhesion kinase and paxillin at residues critical for the regulation of focal adhesion dynamics was diminished in the absence of GEF-H1/RhoA signaling. This study establishes GEF-H1 as a critical organizer of key structural and signaling components of cell migration through the localized regulation of RhoA activity at the cell leading edge.
Figures
Similar articles
-
GEF-H1 couples nocodazole-induced microtubule disassembly to cell contractility via RhoA.Mol Biol Cell. 2008 May;19(5):2147-53. doi: 10.1091/mbc.e07-12-1269. Epub 2008 Feb 20. Mol Biol Cell. 2008. PMID: 18287519 Free PMC article.
-
Polarity-regulating kinase partitioning-defective 1b (PAR1b) phosphorylates guanine nucleotide exchange factor H1 (GEF-H1) to regulate RhoA-dependent actin cytoskeletal reorganization.J Biol Chem. 2011 Dec 30;286(52):44576-84. doi: 10.1074/jbc.M111.267021. Epub 2011 Nov 9. J Biol Chem. 2011. PMID: 22072711 Free PMC article.
-
BNIP-2 retards breast cancer cell migration by coupling microtubule-mediated GEF-H1 and RhoA activation.Sci Adv. 2020 Jul 31;6(31):eaaz1534. doi: 10.1126/sciadv.aaz1534. eCollection 2020 Jul. Sci Adv. 2020. PMID: 32789168 Free PMC article.
-
Regulation and functions of the RhoA regulatory guanine nucleotide exchange factor GEF-H1.Small GTPases. 2021 Sep-Nov;12(5-6):358-371. doi: 10.1080/21541248.2020.1840889. Epub 2020 Oct 30. Small GTPases. 2021. PMID: 33126816 Free PMC article. Review.
-
The guanine nucleotide exchange factor Tiam1: a Janus-faced molecule in cellular signaling.Cell Signal. 2014 Mar;26(3):483-91. doi: 10.1016/j.cellsig.2013.11.034. Epub 2013 Dec 2. Cell Signal. 2014. PMID: 24308970 Review.
Cited by
-
Role of km23-1 in RhoA/actin-based cell migration.Biochem Biophys Res Commun. 2012 Nov 23;428(3):333-8. doi: 10.1016/j.bbrc.2012.10.047. Epub 2012 Oct 15. Biochem Biophys Res Commun. 2012. PMID: 23079622 Free PMC article.
-
The exocyst at the interface between cytoskeleton and membranes in eukaryotic cells.Front Plant Sci. 2014 Jan 2;4:543. doi: 10.3389/fpls.2013.00543. Front Plant Sci. 2014. PMID: 24427163 Free PMC article. Review.
-
TGF-β regulates LARG and GEF-H1 during EMT to affect stiffening response to force and cell invasion.Mol Biol Cell. 2014 Nov 5;25(22):3528-40. doi: 10.1091/mbc.E14-05-1015. Epub 2014 Aug 20. Mol Biol Cell. 2014. PMID: 25143398 Free PMC article.
-
S-1-propenylcysteine improves TNF-α-induced vascular endothelial barrier dysfunction by suppressing the GEF-H1/RhoA/Rac pathway.Cell Commun Signal. 2021 Feb 15;19(1):17. doi: 10.1186/s12964-020-00692-w. Cell Commun Signal. 2021. PMID: 33588881 Free PMC article.
-
Rho GTPase signaling complexes in cell migration and invasion.J Cell Biol. 2018 Feb 5;217(2):447-457. doi: 10.1083/jcb.201612069. Epub 2017 Dec 12. J Cell Biol. 2018. PMID: 29233866 Free PMC article. Review.
References
-
- Baudoin J. P., Alvarez C., Gaspar P., Métin C. Nocodazole-induced changes in microtubule dynamics impair the morphology and directionality of migrating medial ganglionic eminence cells. Dev. Neurosci. 2008;30:132–143. - PubMed
-
- Benais-Pont G., Punn A., Flores-Maldonado C., Eckert J., Raposo G., Fleming T. P., Cereijido M., Balda M. S., Matter K. Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J. Cell Biol. 2003;160:729–740. - PMC - PubMed
-
- Bershadsky A. D., Balaban N. Q., Geiger B. Adhesion-dependent cell mechanosensitivity. Annu. Rev. Cell Dev. Biol. 2003;19:677–695. - PubMed
-
- Brown M. C., Turner C. E. Paxillin: adapting to change. Physiol. Rev. 2004;84:1315–1339. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
