

Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP
- PMID: 19625523
- PMCID: PMC2737362
- DOI: 10.1523/JNEUROSCI.2212-09.2009
Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP
Abstract
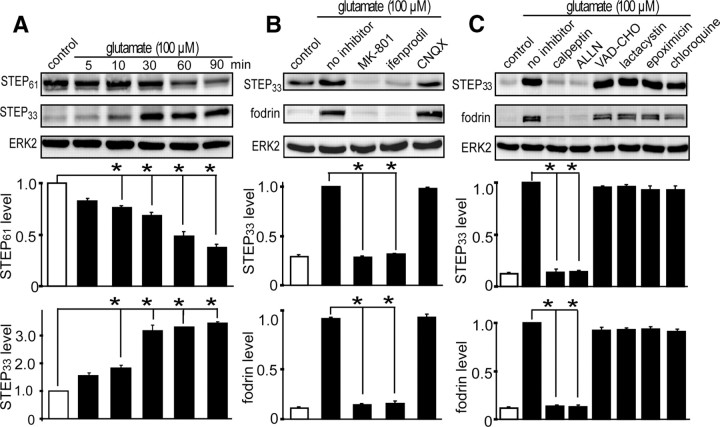
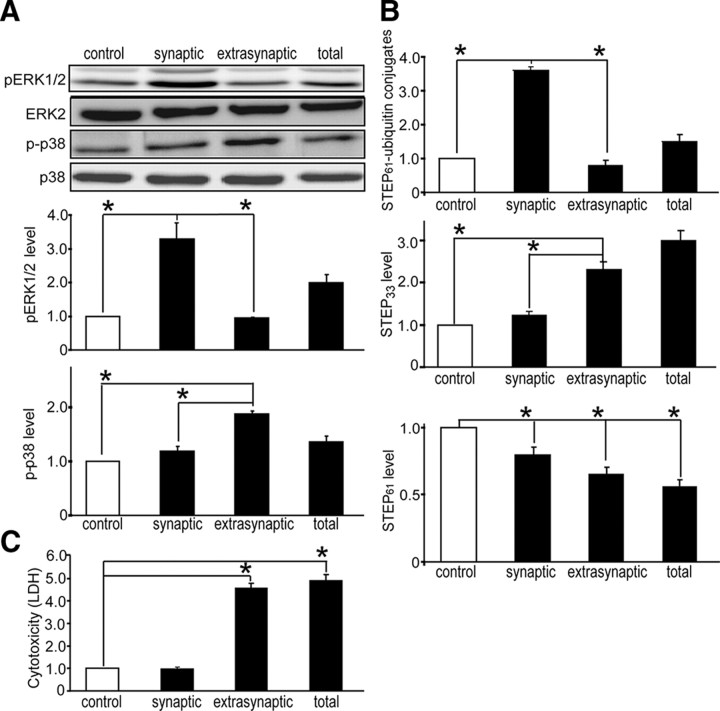
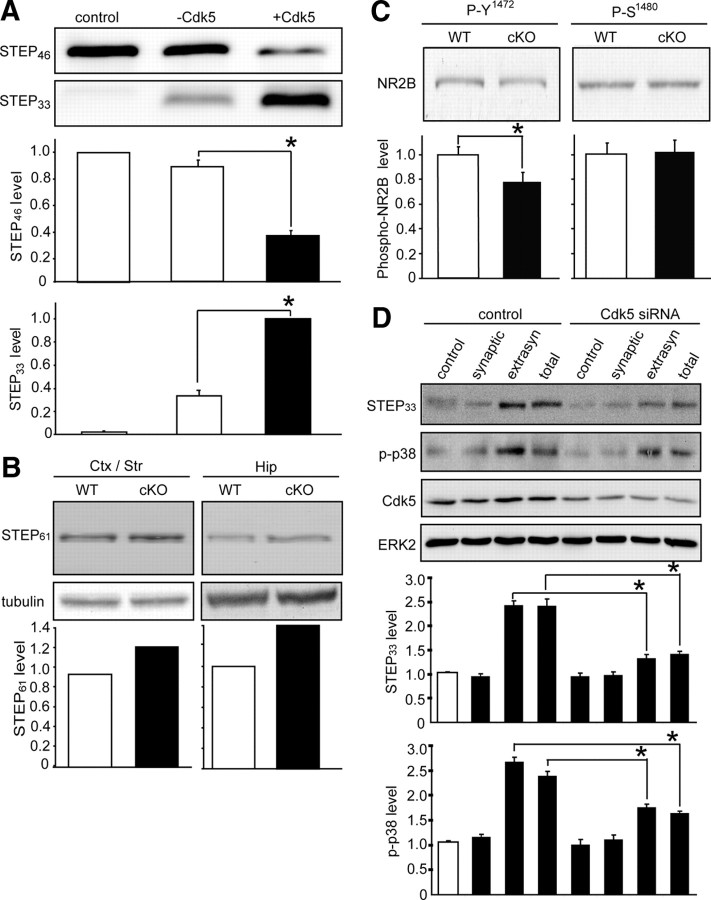
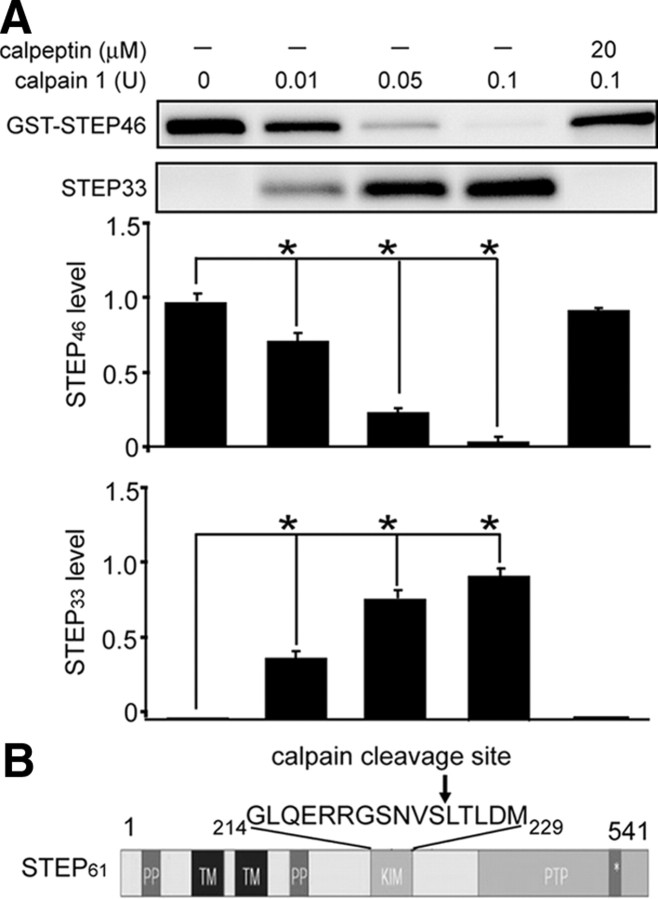
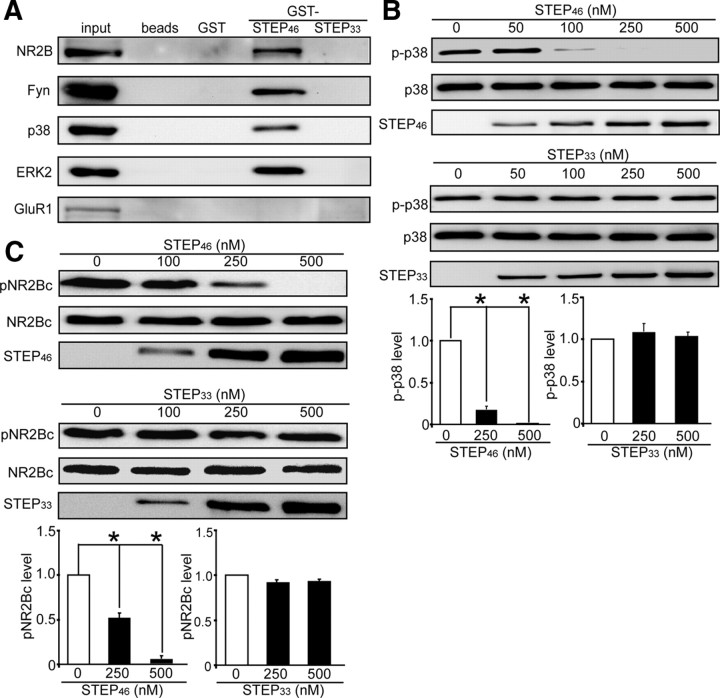
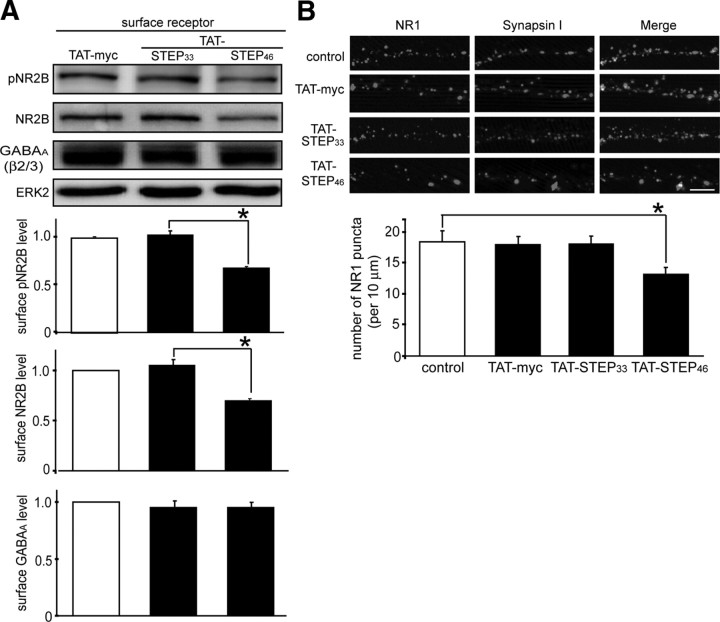
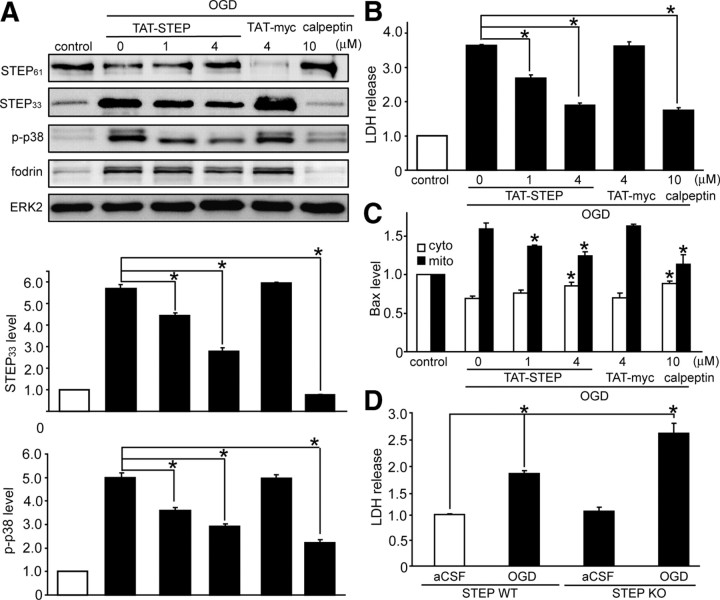
NMDA receptor (NMDAR)-mediated excitotoxicity plays an important role in several CNS disorders, including epilepsy, stroke, and ischemia. Here we demonstrate the involvement of striatal-enriched protein tyrosine phosphatase (STEP) in this critical process. STEP(61) is an alternatively spliced member of the family that is present in postsynaptic terminals. In an apparent paradox, STEP(61) regulates extracellular signal-regulated kinase 1/2 (ERK1/2) and p38, two proteins with opposing functions; activated p38 promotes cell death, whereas activated ERK1/2 promotes cell survival. We found that synaptic stimulation of NMDARs promoted STEP(61) ubiquitination and degradation, concomitant with ERK1/2 activation. In contrast, extrasynaptic stimulation of NMDARs invoked calpain-mediated proteolysis of STEP(61), producing the truncated cleavage product STEP(33) and activation of p38. The calpain cleavage site on STEP was mapped to the kinase interacting motif, a domain required for substrate binding. As a result, STEP(33) neither interacts with nor dephosphorylates STEP substrates. A synthetic peptide spanning the calpain cleavage site efficiently reduced STEP(61) degradation and attenuated p38 activation and cell death in slice models. Furthermore, this peptide was neuroprotective when neurons were subjected to excitotoxicity or cortical slices were exposed to ischemic conditions. These findings suggest a novel mechanism by which differential NMDAR stimulation regulates STEP(61) to promote either ERK1/2 or p38 activation and identifies calpain cleavage of STEP(61) as a valid target for the development of neuroprotective therapy.
Figures

References
-
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298:846–850. - PubMed
-
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. - PubMed
-
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. - PubMed
-
- Besshoh S, Bawa D, Teves L, Wallace MC, Gurd JW. Increased phosphorylation and redistribution of NMDA receptors between synaptic lipid rafts and post-synaptic densities following transient global ischemia in the rat brain. J Neurochem. 2005;93:186–194. - PubMed
-
- Boland B, Campbell V. Beta-amyloid (1–40)-induced apoptosis of cultured cortical neurones involves calpain-mediated cleavage of poly-ADP-ribose polymerase. Neurobiol Aging. 2003;24:179–186. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous