

The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis
- PMID: 19625624
- PMCID: PMC2714280
- DOI: 10.1073/pnas.0906606106
The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis
Abstract
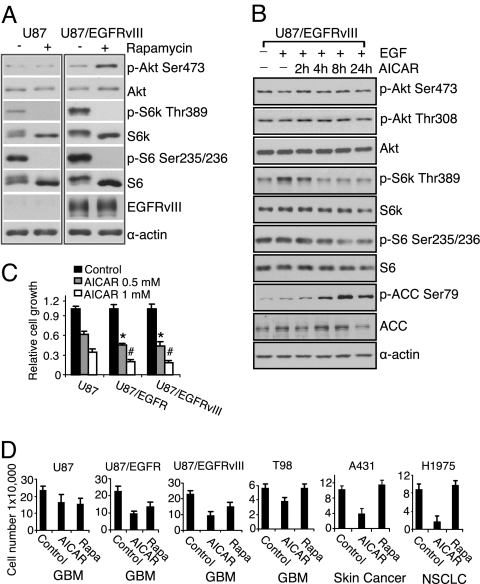
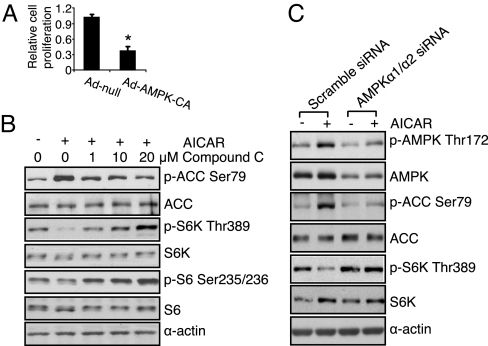
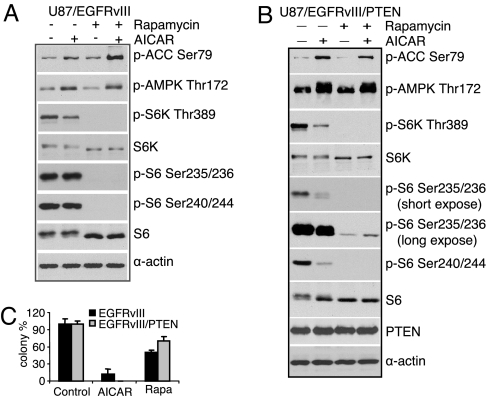
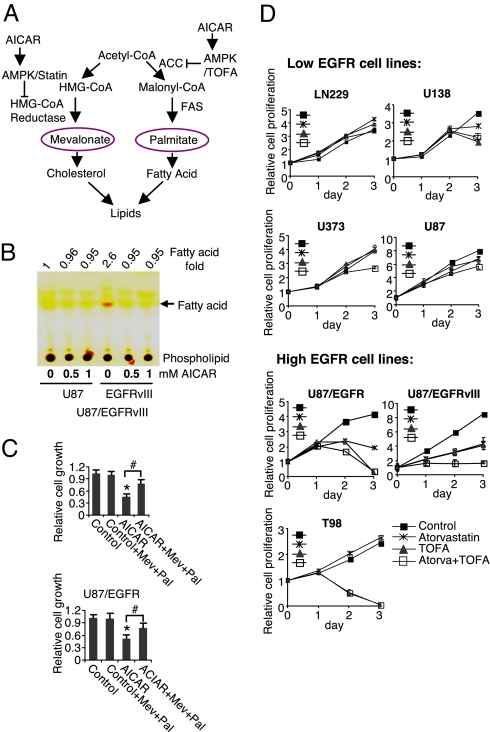
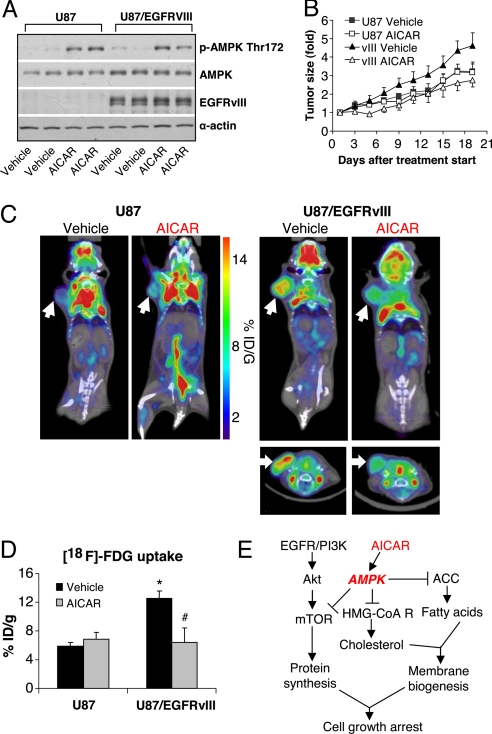
The EGFR/PI3K/Akt/mTOR signaling pathway is activated in many cancers including glioblastoma, yet mTOR inhibitors have largely failed to show efficacy in the clinic. Rapamycin promotes feedback activation of Akt in some patients, potentially underlying clinical resistance and raising the need for alternative approaches to block mTOR signaling. AMPK is a metabolic checkpoint that integrates growth factor signaling with cellular metabolism, in part by negatively regulating mTOR. We used pharmacological and genetic approaches to determine whether AMPK activation could block glioblastoma growth and cellular metabolism, and we examined the contribution of EGFR signaling in determining response in vitro and in vivo. The AMPK-agonist AICAR, and activated AMPK adenovirus, inhibited mTOR signaling and blocked the growth of glioblastoma cells expressing the activated EGFR mutant, EGFRvIII. Across a spectrum of EGFR-activated cancer cell lines, AICAR was more effective than rapamycin at blocking tumor cell proliferation, despite less efficient inhibition of mTORC1 signaling. Unexpectedly, addition of the metabolic products of cholesterol and fatty acid synthesis rescued the growth inhibitory effect of AICAR, whereas inhibition of these lipogenic enzymes mimicked AMPK activation, thus demonstrating that AMPK blocked tumor cell proliferation primarily through inhibition of cholesterol and fatty acid synthesis. Most importantly, AICAR treatment in mice significantly inhibited the growth and glycolysis (as measured by (18)fluoro-2-deoxyglucose microPET) of glioblastoma xenografts engineered to express EGFRvIII, but not their parental counterparts. These results suggest a mechanism by which AICAR inhibits the proliferation of EGFRvIII expressing glioblastomas and point toward a potential therapeutic strategy for targeting EGFR-activated cancers.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Yap TA, et al. Targeting the PI3K-AKT-mTOR pathway: Progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. - PubMed
-
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. - PubMed
-
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signaling controls tumor cell growth. Nature. 2006;441:424–430. - PubMed
-
- Choe G, et al. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63:2742–2746. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
