

Thrombomodulin mutations in atypical hemolytic-uremic syndrome
- PMID: 19625716
- PMCID: PMC3530919
- DOI: 10.1056/NEJMoa0810739
Thrombomodulin mutations in atypical hemolytic-uremic syndrome
Abstract
Background: The hemolytic-uremic syndrome consists of the triad of microangiopathic hemolytic anemia, thrombocytopenia, and renal failure. The common form of the syndrome is triggered by infection with Shiga toxin-producing bacteria and has a favorable outcome. The less common form of the syndrome, called atypical hemolytic-uremic syndrome, accounts for about 10% of cases, and patients with this form of the syndrome have a poor prognosis. Approximately half of the patients with atypical hemolytic-uremic syndrome have mutations in genes that regulate the complement system. Genetic factors in the remaining cases are unknown. We studied the role of thrombomodulin, an endothelial glycoprotein with anticoagulant, antiinflammatory, and cytoprotective properties, in atypical hemolytic-uremic syndrome.
Methods: We sequenced the entire thrombomodulin gene (THBD) in 152 patients with atypical hemolytic-uremic syndrome and in 380 controls. Using purified proteins and cell-expression systems, we investigated whether thrombomodulin regulates the complement system, and we characterized the mechanisms. We evaluated the effects of thrombomodulin missense mutations associated with atypical hemolytic-uremic syndrome on complement activation by expressing thrombomodulin variants in cultured cells.
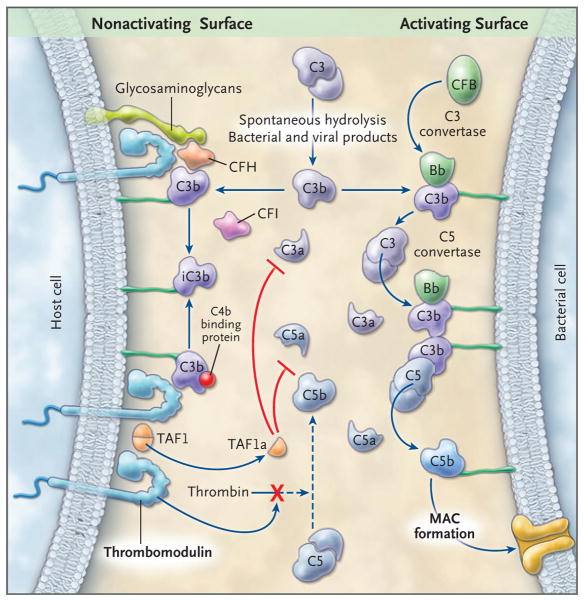
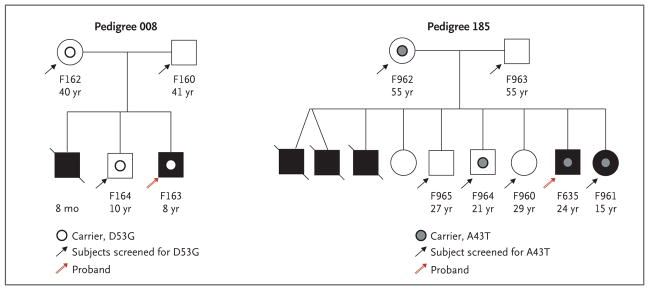
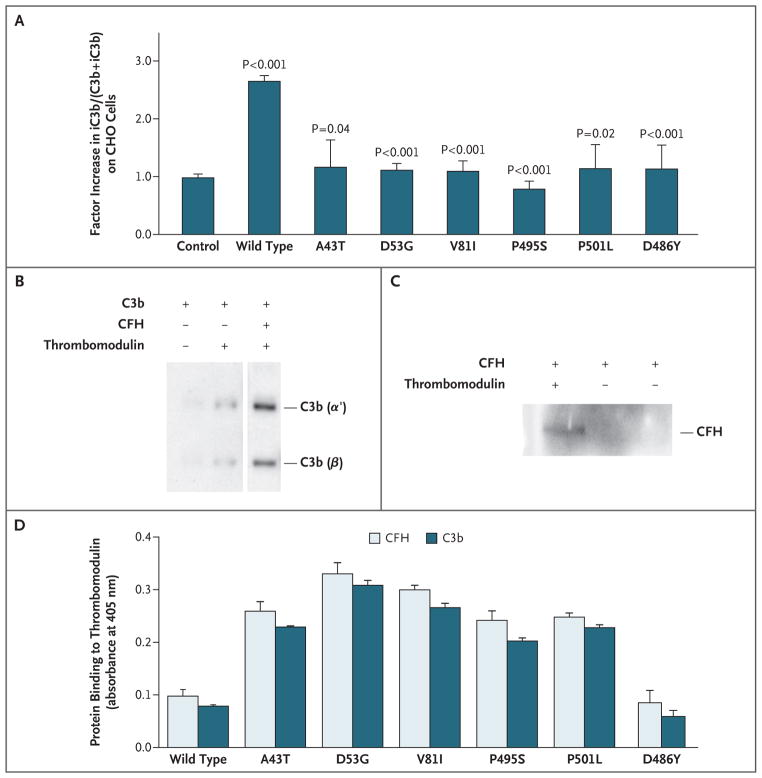
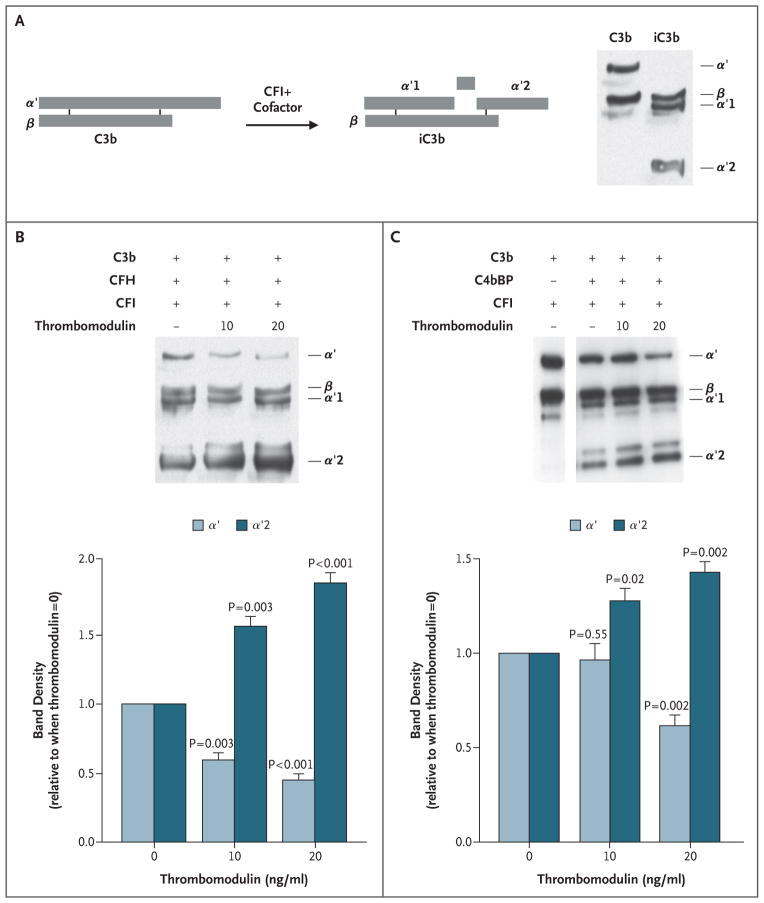
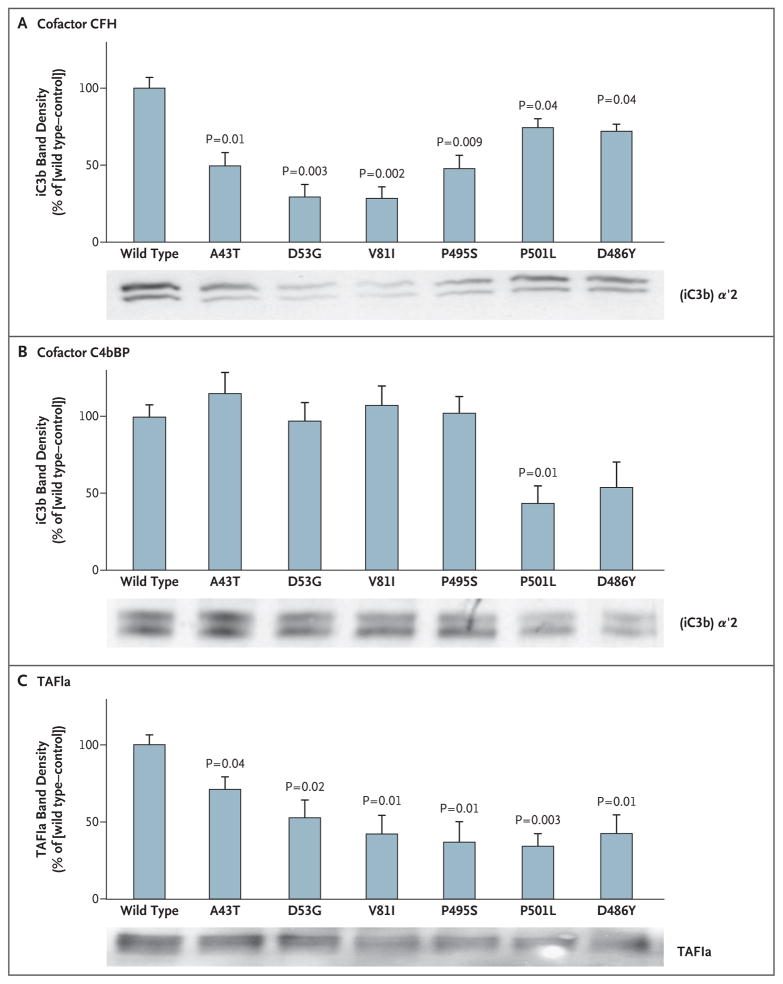
Results: Of 152 patients with atypical hemolytic-uremic syndrome, 7 unrelated patients had six different heterozygous missense THBD mutations. In vitro, thrombomodulin binds to C3b and factor H (CFH) and negatively regulates complement by accelerating factor I-mediated inactivation of C3b in the presence of cofactors, CFH or C4b binding protein. By promoting activation of the plasma procarboxypeptidase B, thrombomodulin also accelerates the inactivation of anaphylatoxins C3a and C5a. Cultured cells expressing thrombomodulin variants associated with atypical hemolytic-uremic syndrome had diminished capacity to inactivate C3b and to activate procarboxypeptidase B and were thus less protected from activated complement.
Conclusions: Mutations that impair the function of thrombomodulin occur in about 5% of patients with atypical hemolytic-uremic syndrome.
2009 Massachusetts Medical Society
Conflict of interest statement
Drs. C.T. Esmon and N.L. Esmon report holding licenses and patents related to protein C and activated protein C that are unrelated to this article; and Dr. Conway, holding a patent for the use of the lectinlike domain of thrombomodulin as an antiinflammatory agent. No other potential conflict of interest relevant to this article was reported.
Figures
Comment in
-
Thrombomodulin in atypical hemolytic-uremic syndrome.N Engl J Med. 2009 Oct 8;361(15):1511; author reply 1511. doi: 10.1056/NEJMc091704. N Engl J Med. 2009. PMID: 19812413 No abstract available.
References
-
- Constantinescu AR, Bitzan M, Weiss LS, et al. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis. 2004;43:976–82. - PubMed
-
- Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589–600. - PubMed
-
- Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:1035–50. - PubMed
-
- Razzaq S. Hemolytic uremic syndrome: an emerging health risk. Am Fam Physician. 2006;74:991–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous