

False discovery rates of protein identifications: a strike against the two-peptide rule
- PMID: 19627159
- PMCID: PMC3398614
- DOI: 10.1021/pr9004794
False discovery rates of protein identifications: a strike against the two-peptide rule
Abstract
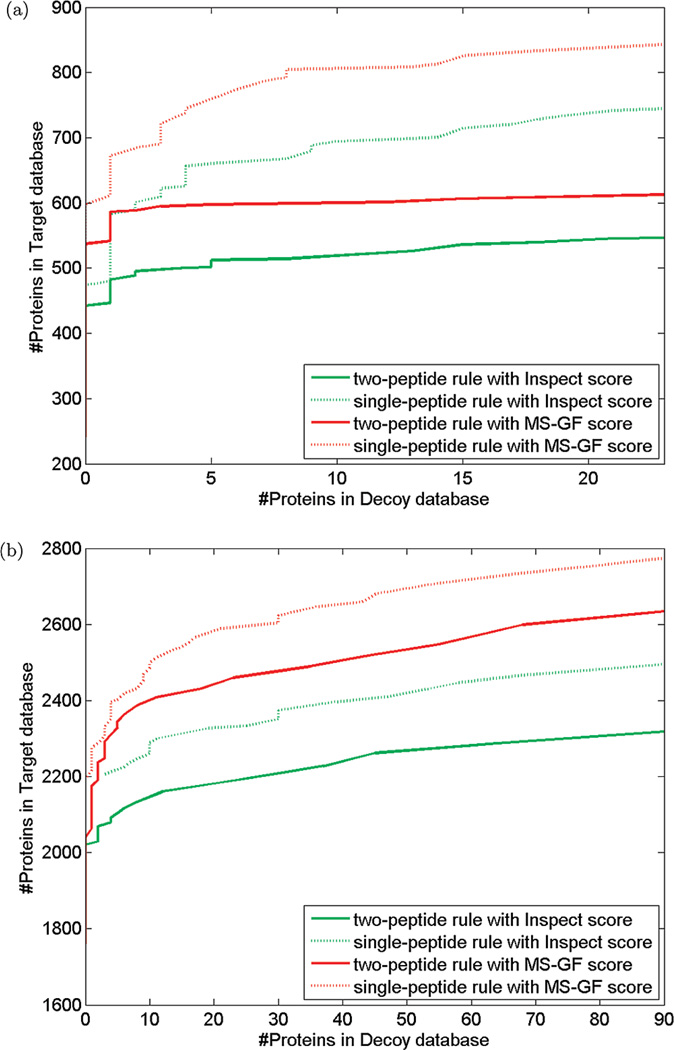
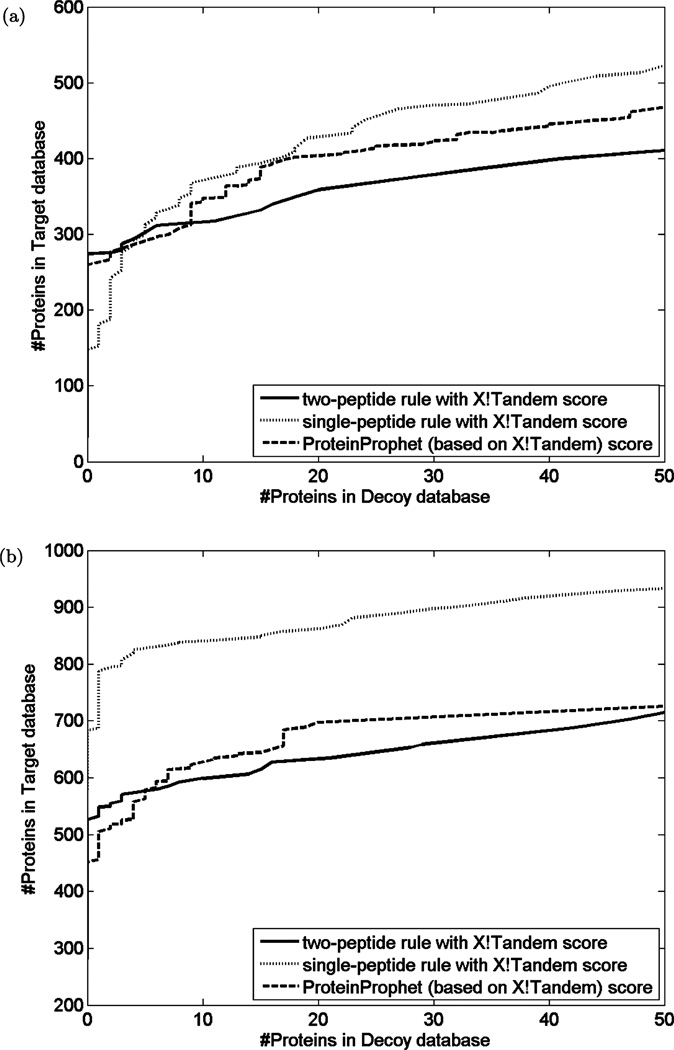
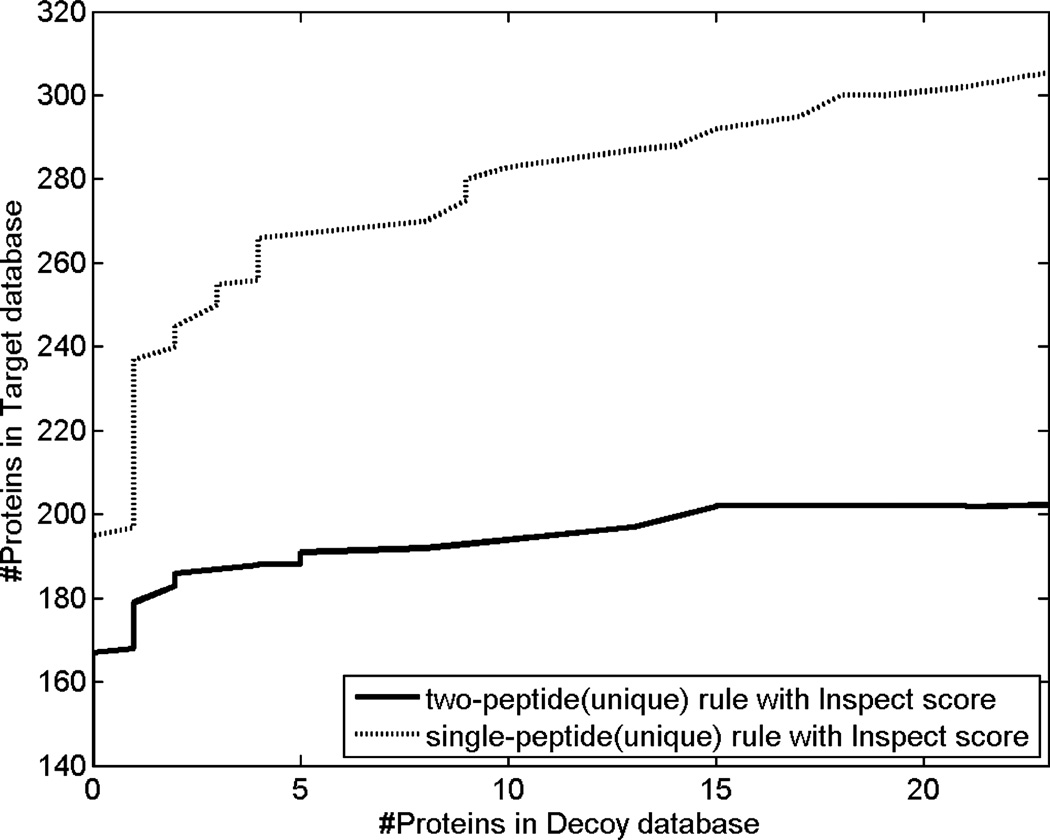
Most proteomics studies attempt to maximize the number of peptide identifications and subsequently infer proteins containing two or more peptides as reliable protein identifications. In this study, we evaluate the effect of this "two-peptide" rule on protein identifications, using multiple search tools and data sets. Contrary to the intuition, the "two-peptide" rule reduces the number of protein identifications in the target database more significantly than in the decoy database and results in increased false discovery rates, compared to the case when single-hit proteins are not discarded. We therefore recommend that the "two-peptide" rule should be abandoned, and instead, protein identifications should be subject to the estimation of error rates, as is the case with peptide identifications. We further extend the generating function approach (originally proposed for evaluating matches between a peptide and a single spectrum) to evaluating matches between a protein and an entire spectral data set.
Figures


References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Cargile BJ, Bundy JL, Stephenson JL., Jr Potential for false positive identifications from large databases through tandem mass spectrometry. J. Proteome Res. 2004;3:1082–1085. - PubMed
-
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–214. - PubMed
-
- Kall L, Storey JD, MacCoss MJ, Noble SW. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 2008;7:29–34. - PubMed
-
- Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, Apweiler R, Haab BB, Simpson RJ, Eddes JS. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5:3226–3245. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
