

9-Aminoacridine inhibition of HIV-1 Tat dependent transcription
- PMID: 19630958
- PMCID: PMC2723079
- DOI: 10.1186/1743-422X-6-114
9-Aminoacridine inhibition of HIV-1 Tat dependent transcription
Abstract
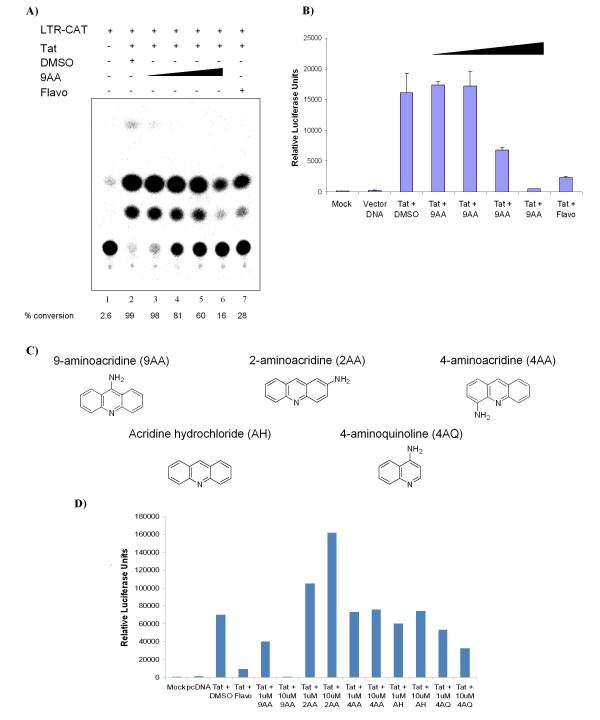
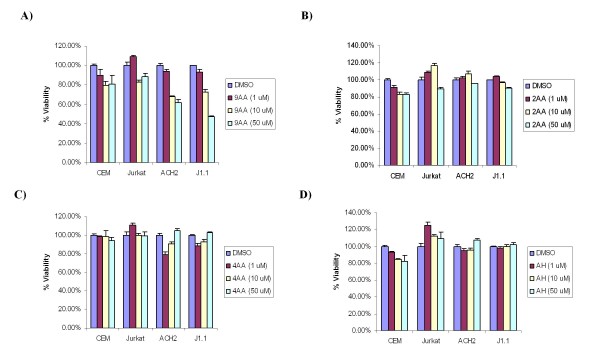
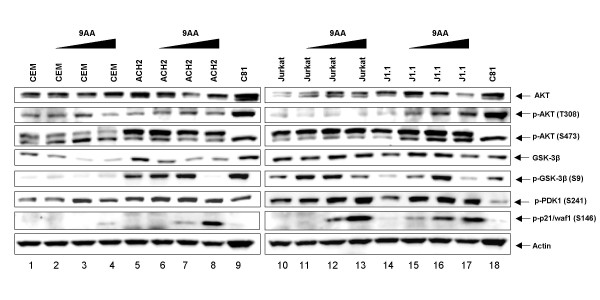
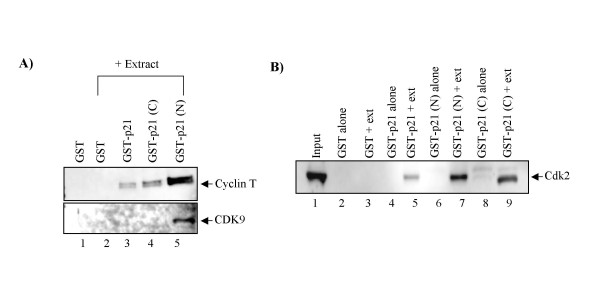
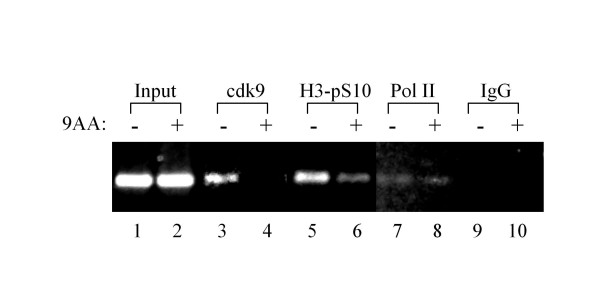
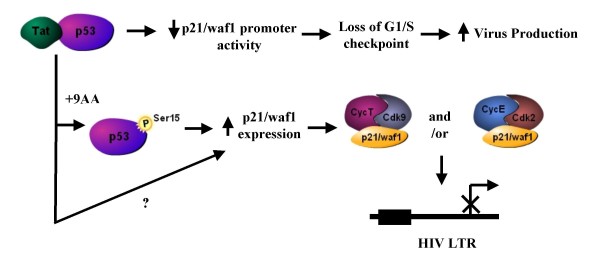
As part of a continued search for more efficient anti-HIV-1 drugs, we are focusing on the possibility that small molecules could efficiently inhibit HIV-1 replication through the restoration of p53 and p21WAF1 functions, which are inactivated by HIV-1 infection. Here we describe the molecular mechanism of 9-aminoacridine (9AA) mediated HIV-1 inhibition. 9AA treatment resulted in inhibition of HIV LTR transcription in a specific manner that was highly dependent on the presence and location of the amino moiety. Importantly, virus replication was found to be inhibited in HIV-1 infected cell lines by 9AA in a dose-dependent manner without inhibiting cellular proliferation or inducing cell death. 9AA inhibited viral replication in both p53 wildtype and p53 mutant cells, indicating that there is another p53 independent factor that was critical for HIV inhibition. p21WAF1 is an ideal candidate as p21WAF1 levels were increased in both p53 wildtype and p53 mutant cells, and p21WAF1 was found to be phosphorylated at S146, an event previously shown to increase its stability. Furthermore, we observed p21WAF1 in complex with cyclin T1 and cdk9 in vitro, suggesting a direct role of p21WAF1 in HIV transcription inhibition. Finally, 9AA treatment resulted in loss of cdk9 from the viral promoter, providing one possible mechanism of transcriptional inhibition. Thus, 9AA treatment was highly efficient at reactivating the p53 - p21WAF1 pathway and consequently inhibiting HIV replication and transcription.
Figures

Similar articles
-
Drug 9AA reactivates p21/Waf1 and Inhibits HIV-1 progeny formation.Virol J. 2008 Mar 18;5:41. doi: 10.1186/1743-422X-5-41. Virol J. 2008. PMID: 18348731 Free PMC article.
-
Combination of 9-aminoacridine with Campath-1H provides effective therapy for a murine model of adult T-cell leukemia.Retrovirology. 2014 Jun 2;11:43. doi: 10.1186/1742-4690-11-43. Retrovirology. 2014. PMID: 24890041 Free PMC article.
-
Specific Inhibition of HIV Infection by the Action of Spironolactone in T Cells.J Virol. 2016 Nov 14;90(23):10972-10980. doi: 10.1128/JVI.01722-16. Print 2016 Dec 1. J Virol. 2016. PMID: 27681137 Free PMC article.
-
Inhibitors of HIV-1 gene expression and transcription.Curr Top Med Chem. 2004;4(9):871-82. doi: 10.2174/1568026043388466. Curr Top Med Chem. 2004. PMID: 15134546 Review.
-
Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure.Viruses. 2020 Apr 8;12(4):415. doi: 10.3390/v12040415. Viruses. 2020. PMID: 32276443 Free PMC article. Review.
Cited by
-
microRNA machinery is an integral component of drug-induced transcription inhibition in HIV-1 infection.J RNAi Gene Silencing. 2010 May 29;6(1):386-400. J RNAi Gene Silencing. 2010. PMID: 20628499 Free PMC article.
-
Strategies to Block HIV Transcription: Focus on Small Molecule Tat Inhibitors.Biology (Basel). 2012 Nov 19;1(3):668-97. doi: 10.3390/biology1030668. Biology (Basel). 2012. PMID: 24832514 Free PMC article.
-
Eradication of HIV and Cure of AIDS, Now and How?Front Immunol. 2013 Oct 18;4:337. doi: 10.3389/fimmu.2013.00337. Front Immunol. 2013. PMID: 24151495 Free PMC article. Review.
-
Role of p53 in neurodegenerative diseases.Neurodegener Dis. 2012;9(2):68-80. doi: 10.1159/000329999. Epub 2011 Oct 28. Neurodegener Dis. 2012. PMID: 22042001 Free PMC article. Review.
-
Role of cellular iron and oxygen in the regulation of HIV-1 infection.Future Virol. 2013 Mar;8(3):301-311. doi: 10.2217/fvl.13.6. Future Virol. 2013. PMID: 23678366 Free PMC article.
References
-
- Liu Y, Kulesz-Martin M. P53 regulation and function in normal cells and tumors. Medicina (B Aires) 2000;60:9–11. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
