

Redox control of asthma: molecular mechanisms and therapeutic opportunities
- PMID: 19634987
- PMCID: PMC2824520
- DOI: 10.1089/ars.2008.2425
Redox control of asthma: molecular mechanisms and therapeutic opportunities
Erratum in
- Antioxid Redox Signal. 2010 Feb;12(2):321. Ghio,Andrew [removed]; Kinnula, Vuokko [removed]; Kliment, Corrine [removed];Montuschi, Paolo [removed]; Reddy, Sekhar [removed]; White, Carl [removed]
Abstract
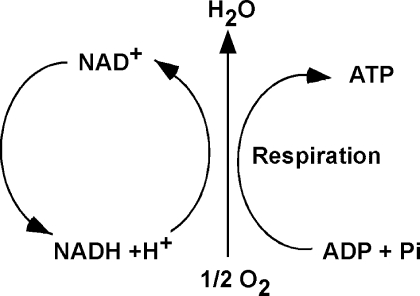
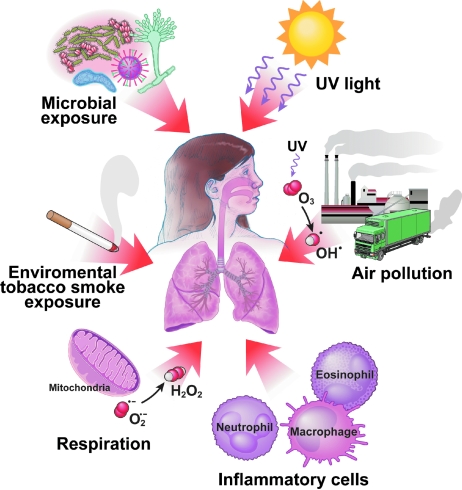
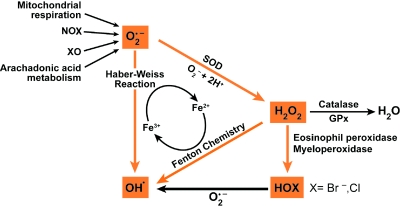
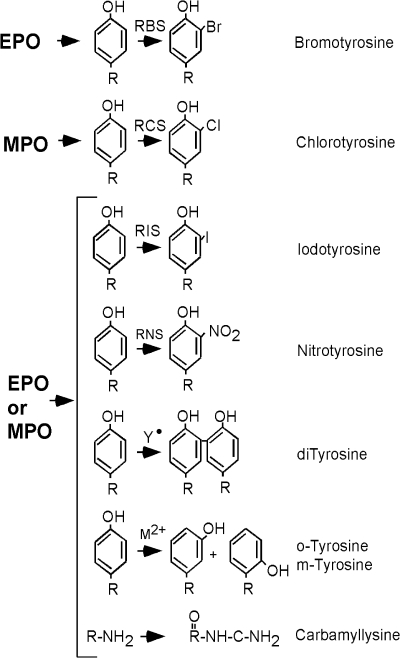
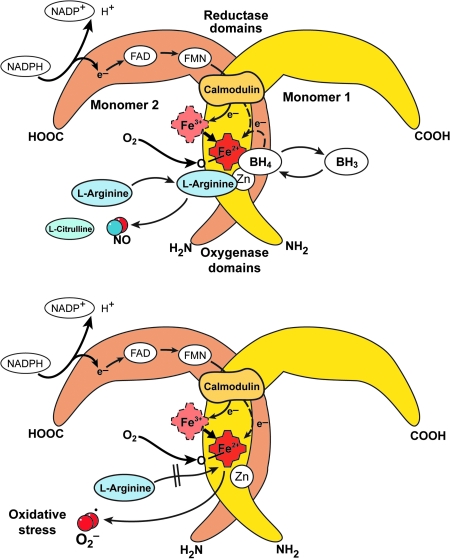
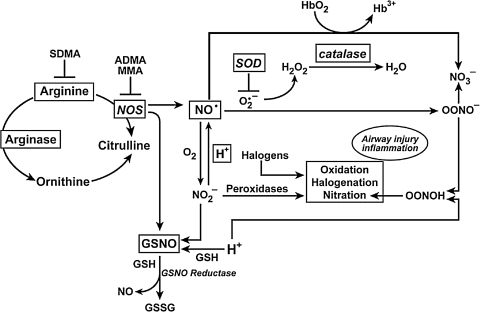
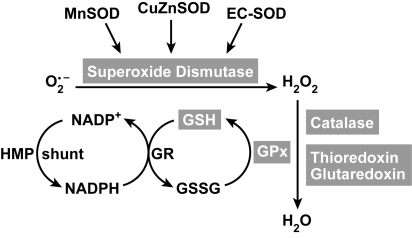
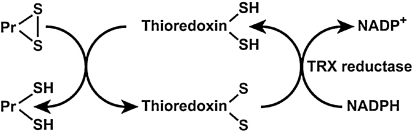
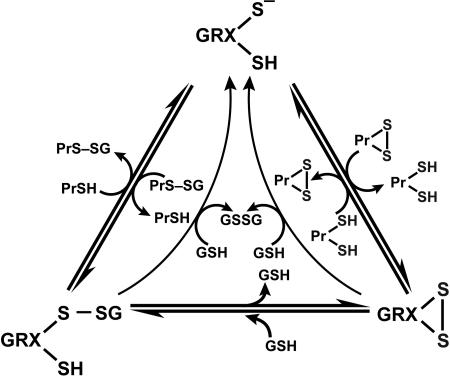
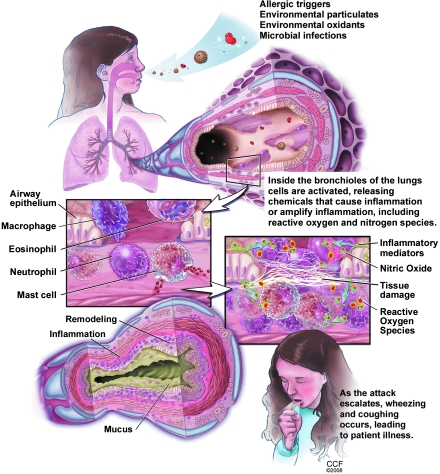
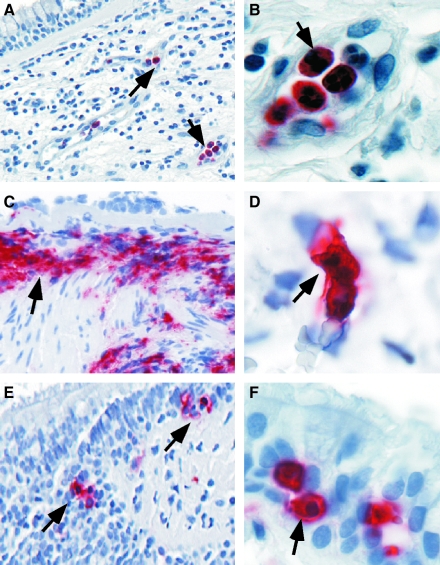
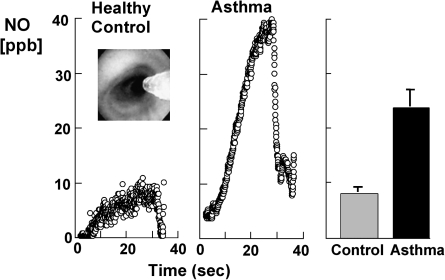
An imbalance in reducing and oxidizing (redox) systems favoring a more oxidative environment is present in asthma and linked to the pathophysiology of the defining symptoms and signs including airflow limitation, hyper-reactivity, and airway remodeling. High levels of hydrogen peroxide, nitric oxide ((*)NO), and 15-F(2t)-isoprostane in exhaled breath, and excessive oxidative protein products in lung epithelial lining fluid, peripheral blood, and urine provide abundant evidence for pathologic oxidizing processes in asthma. Parallel studies document loss of reducing potential by nonenzymatic and enzymatic antioxidants. The essential first line antioxidant enzymes superoxide dismutases (SOD) and catalase are reduced in asthma as compared to healthy individuals, with lowest levels in those patients with the most severe asthma. Loss of SOD and catalase activity is related to oxidative modifications of the enzymes, while other antioxidant gene polymorphisms are linked to susceptibility to develop asthma. Monitoring of exhaled (*)NO has entered clinical practice because it is useful to optimize asthma care, and a wide array of other biochemical oxidative and nitrative biomarkers are currently being evaluated for asthma monitoring and phenotyping. Novel therapeutic strategies that target correction of redox abnormalities show promise for the treatment of asthma.
Figures
References
-
- Abu-Soud HM. Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. J Biol Chem. 2000;275:37524–37532. - PubMed
-
- Abu-Soud HM. Hazen SL. Nitric oxide modulates the catalytic activity of myeloperoxidase. J Biol Chem. 2000;275:5425–5430. - PubMed
-
- Abu-Soud HM. Khassawneh MY. Sohn JT. Murray P. Haxhiu MA. Hazen SL. Peroxidases inhibit nitric oxide (NO) dependent bronchodilation: Development of a model describing NO-peroxidase interactions. Biochemistry. 2001;40:11866–11875. - PubMed
-
- Adcock IM. Brown CR. Kwon O. Barnes PJ. Oxidative stress induces NF kappa B DNA binding and inducible NOS mRNA in human epithelial cells. Biochem Biophys Res Commun. 1994;199:1518–1524. - PubMed
-
- Agani FH. Pichiule P. Chavez JC. LaManna JC. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J Biol Chem. 2000;275:35863–35867. - PubMed
