

The external pore loop interacts with S6 and S3-S4 linker in domain 4 to assume an essential role in gating control and anticonvulsant action in the Na(+) channel
- PMID: 19635852
- PMCID: PMC2717694
- DOI: 10.1085/jgp.200810158
The external pore loop interacts with S6 and S3-S4 linker in domain 4 to assume an essential role in gating control and anticonvulsant action in the Na(+) channel
Abstract
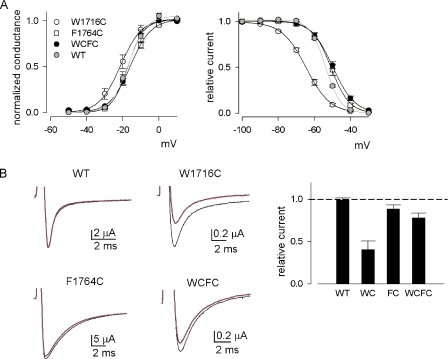
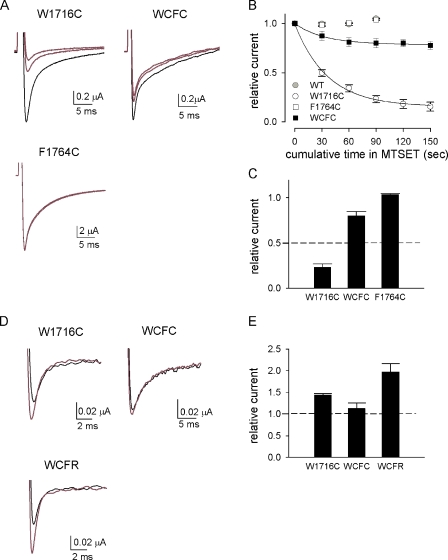
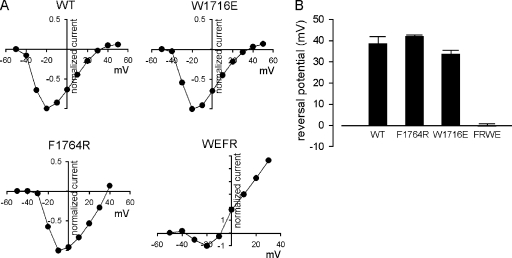
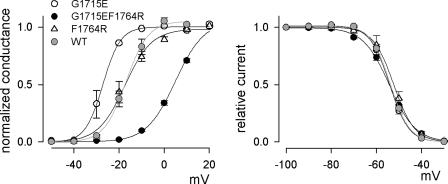
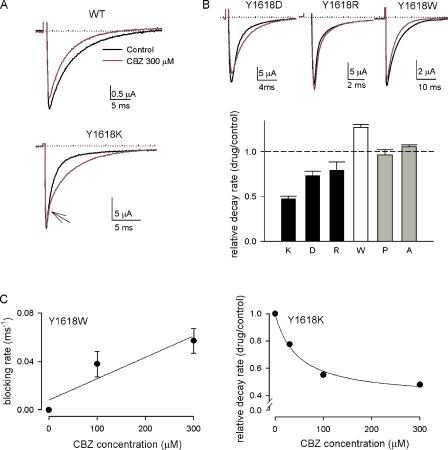
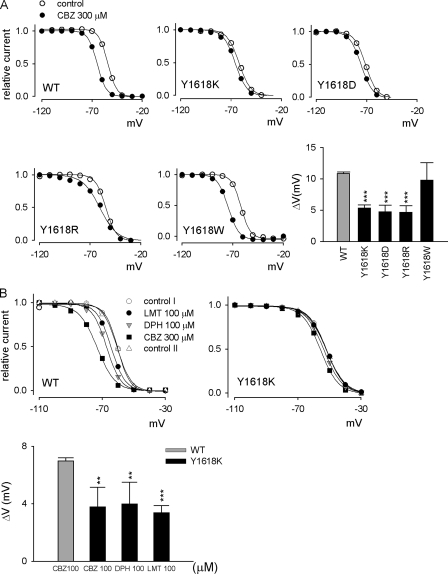
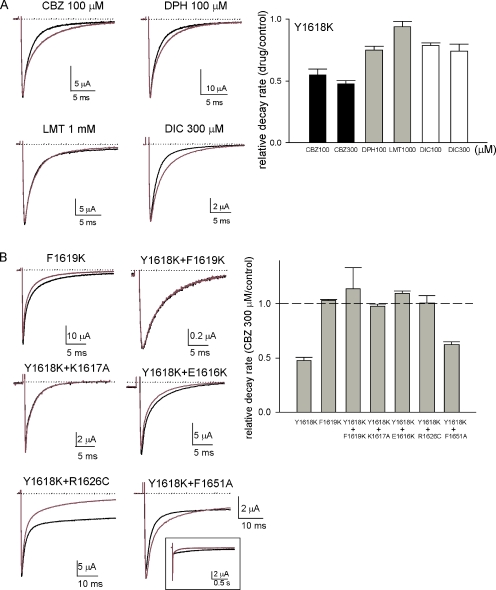
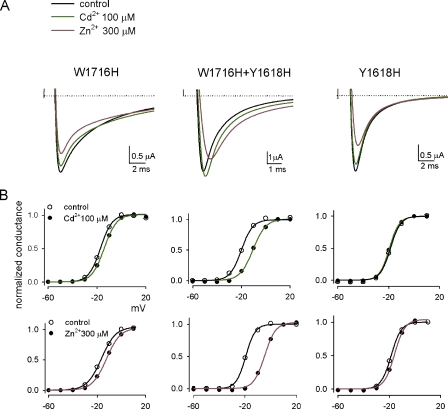
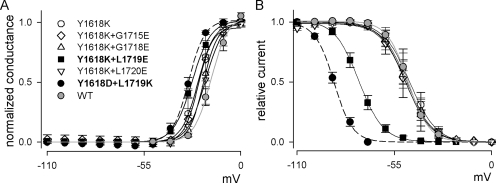
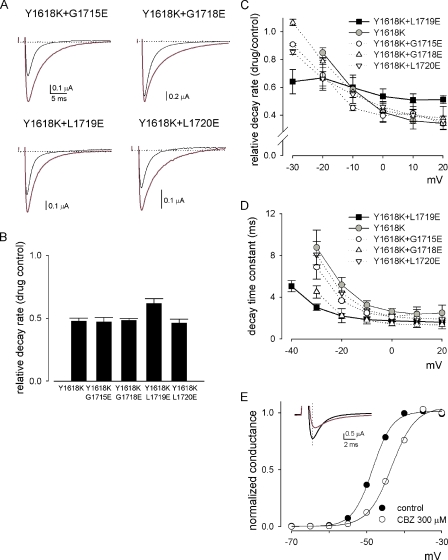
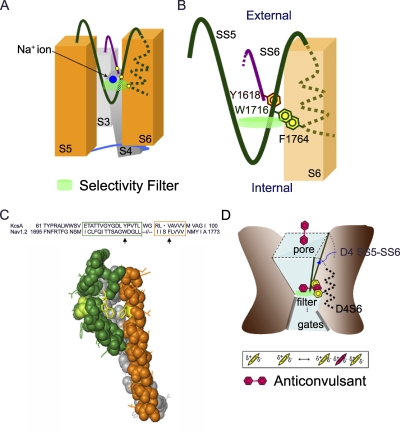
Carbamazepine, phenytoin, and lamotrigine are widely prescribed anticonvulsants in neurological clinics. These drugs bind to the same receptor site, probably with the diphenyl motif in their structure, to inhibit the Na(+) channel. However, the location of the drug receptor remains controversial. In this study, we demonstrate close proximity and potential interaction between an external aromatic residue (W1716 in the external pore loop) and an internal aromatic residue (F1764 in the pore-lining part of the sixth transmembrane segment, S6) of domain 4 (D4), both being closely related to anticonvulsant and/or local anesthetic binding to the Na(+) channel. Double-mutant cycle analysis reveals significant cooperativity between the two phenyl residues for anticonvulsant binding. Concomitant F1764C mutation evidently decreases the susceptibility of W1716C to external Cd(2+) and membrane-impermeable methanethiosulfonate reagents. Also, the W1716E/F1764R and G1715E/F1764R double mutations significantly alter the selectivity for Na(+) over K(+) and markedly shift the activation curve, respectively. W1716 and F1764 therefore very likely form a link connecting the outer and inner compartments of the Na(+) channel pore (in addition to the selectivity filter). Anticonvulsants and local anesthetics may well traverse this "S6 recess" without trespassing on the selectivity filter. Furthermore, we found that Y1618K, a point mutation in the S3-4 linker (the extracellular extension of D4S4), significantly alters the consequences of carbamazepine binding to the Na(+) channel. The effect of Y1618K mutation, however, is abolished by concomitant point mutations in the vicinity of Y1618, but not by those in the internally located inactivation machinery, supporting a direct local rather than a long-range allosteric action. Moreover, Y1618 could interact with D4 pore residues W1716 and L1719 to have a profound effect on both channel gating and anticonvulsant action. We conclude that there are direct interactions among the external S3-4 linker, the external pore loop, and the internal S6 segment in D4, making the external pore loop a pivotal point critically coordinating ion permeation, gating, and anticonvulsant binding in the Na(+) channel.
Figures
Similar articles
-
Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit.J Biol Chem. 2001 Jan 5;276(1):20-7. doi: 10.1074/jbc.M006992200. J Biol Chem. 2001. PMID: 11024055
-
A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels.Mol Pharmacol. 1998 Oct;54(4):712-21. Mol Pharmacol. 1998. PMID: 9765515
-
Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore.Mol Pharmacol. 2010 Oct;78(4):631-8. doi: 10.1124/mol.110.064683. Epub 2010 Jul 19. Mol Pharmacol. 2010. PMID: 20643904 Free PMC article.
-
Molecular properties of brain sodium channels: an important target for anticonvulsant drugs.Adv Neurol. 1999;79:441-56. Adv Neurol. 1999. PMID: 10514834 Review.
-
Molecular pore structure of voltage-gated sodium and calcium channels.Braz J Med Biol Res. 1994 Dec;27(12):2781-802. Braz J Med Biol Res. 1994. PMID: 7550000 Review.
Cited by
-
A molecular switch between the outer and the inner vestibules of the voltage-gated Na+ channel.J Biol Chem. 2010 Dec 10;285(50):39458-70. doi: 10.1074/jbc.M110.132886. Epub 2010 Oct 6. J Biol Chem. 2010. PMID: 20926383 Free PMC article.
-
Gating transitions in the selectivity filter region of a sodium channel are coupled to the domain IV voltage sensor.Proc Natl Acad Sci U S A. 2012 Feb 14;109(7):2648-53. doi: 10.1073/pnas.1210413109. Epub 2012 Jan 30. Proc Natl Acad Sci U S A. 2012. PMID: 22308389 Free PMC article.
-
Fast- or slow-inactivated state preference of Na+ channel inhibitors: a simulation and experimental study.PLoS Comput Biol. 2010 Jun 17;6(6):e1000818. doi: 10.1371/journal.pcbi.1000818. PLoS Comput Biol. 2010. PMID: 20585544 Free PMC article.
-
A naturally occurring amino acid substitution in the voltage-dependent sodium channel selectivity filter affects channel gating.J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2013 Oct;199(10):829-42. doi: 10.1007/s00359-013-0845-3. Epub 2013 Aug 25. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2013. PMID: 23979192
-
Kinetic Alterations in Resurgent Sodium Currents of Mutant Nav1.4 Channel in Two Patients Affected by Paramyotonia Congenita.Biology (Basel). 2022 Apr 18;11(4):613. doi: 10.3390/biology11040613. Biology (Basel). 2022. PMID: 35453812 Free PMC article.
References
-
- Benzinger G.R., Kyle J.W., Blumenthal K.M., Hanck D.A. 1998. A specific interaction between the cardiac sodium channel and site-3 toxin anthopleurin B.J. Biol. Chem. 273:80–84 - PubMed
-
- Butterworth J.F., Strichartz G.R. 1990. Molecular mechanisms of local anesthesia: a review.Anesthesiology. 72:711–734 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
