

Second-by-second analysis of alpha 7 nicotine receptor regulation of glutamate release in the prefrontal cortex of awake rats
- PMID: 19637277
- PMCID: PMC2759414
- DOI: 10.1002/syn.20693
Second-by-second analysis of alpha 7 nicotine receptor regulation of glutamate release in the prefrontal cortex of awake rats
Abstract
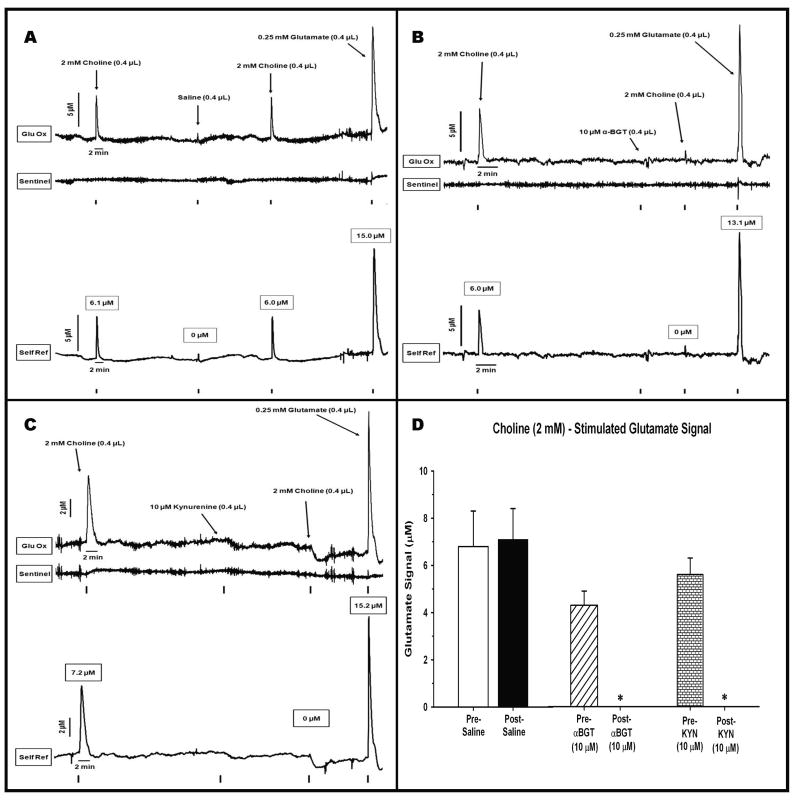
These experiments utilized an enzyme-based microelectrode selective for the second-by-second detection of extracellular glutamate to reveal the alpha 7-based nicotinic modulation of glutamate release in the prefrontal cortex (PFC) of freely moving rats. Rats received intracortical infusions of the nonselective nicotinic agonist nicotine (12.0 mM, 1.0 microg/0.4 microl) or the selective alpha 7 agonist choline (2.0 mM/0.4 microl). The selectivity of drug-induced glutamate release was assessed in subgroups of animals pretreated with the alpha 7 antagonist, alpha-bungarotoxin (alpha-BGT, 10 microM), or kynurenine (10 microM) the precursor of the astrocyte-derived, negative allosteric alpha 7 modulator kynurenic acid. Local administration of nicotine increased glutamate signals (maximum amplitude = 4.3 +/- 0.6 microM) that were cleared to baseline levels in 493 +/- 80 seconds. Pretreatment with alpha-BGT or kynurenine attenuated nicotine-induced glutamate by 61% and 60%, respectively. Local administration of choline also increased glutamate signals (maximum amplitude = 6.3 +/- 0.9 microM). In contrast to nicotine-evoked glutamate release, choline-evoked signals were cleared more quickly (28 +/- 6 seconds) and pretreatment with alpha-BGT or kynurenine completely blocked the stimulated glutamate release. Using a method that reveals the temporal dynamics of in vivo glutamate release and clearance, these data indicate a nicotinic modulation of cortical glutamate release that is both alpha 7- and non-alpha 7-mediated. Furthermore, these data may also provide a mechanism underlying the recent focus on alpha 7 full and partial agonists as therapeutic agents in the treatment of cortically mediated cognitive deficits in schizophrenia.
(c) 2009 Wiley-Liss, Inc.
Figures
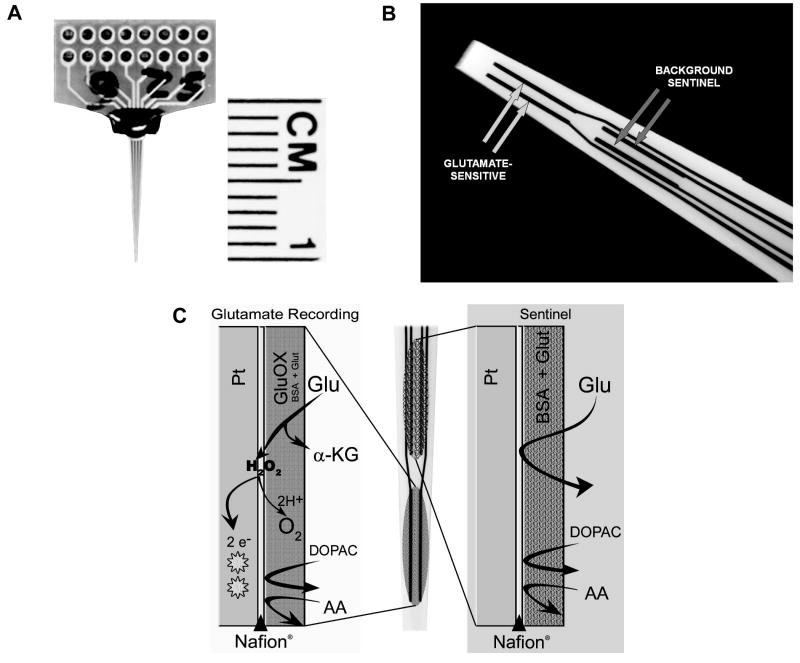
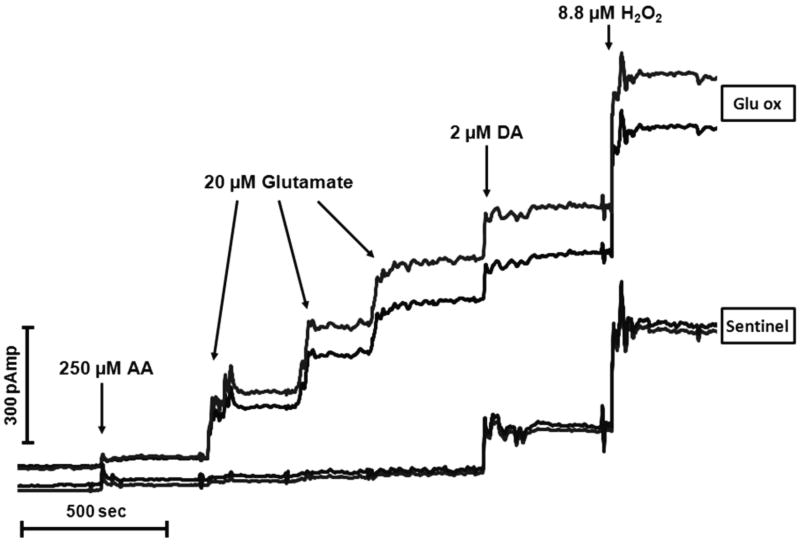
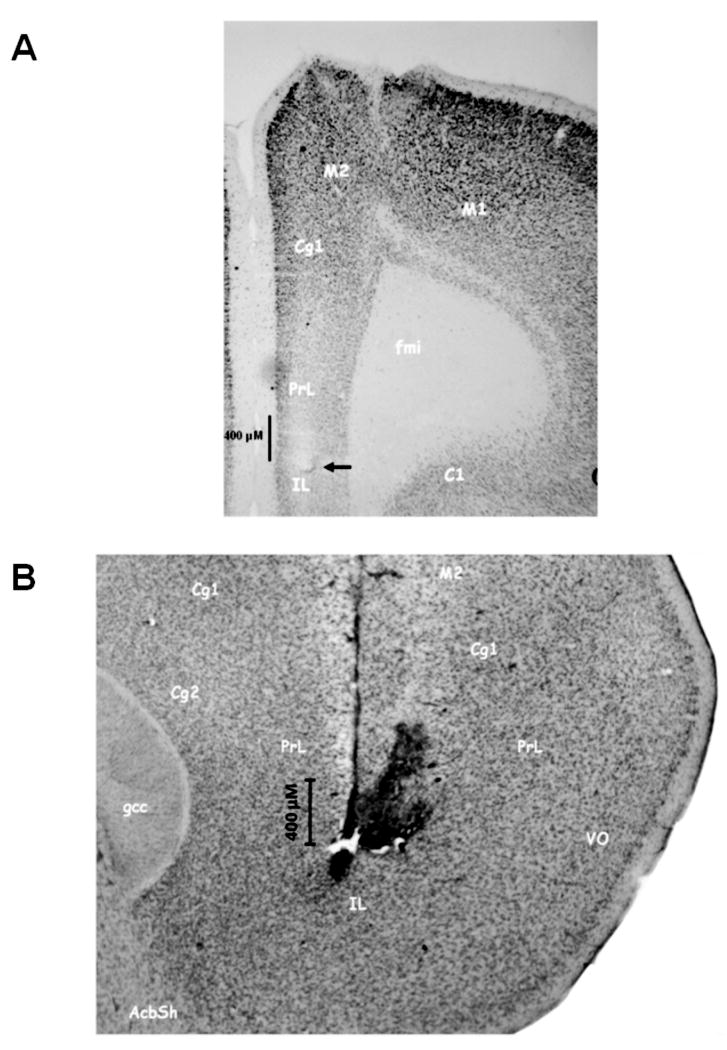
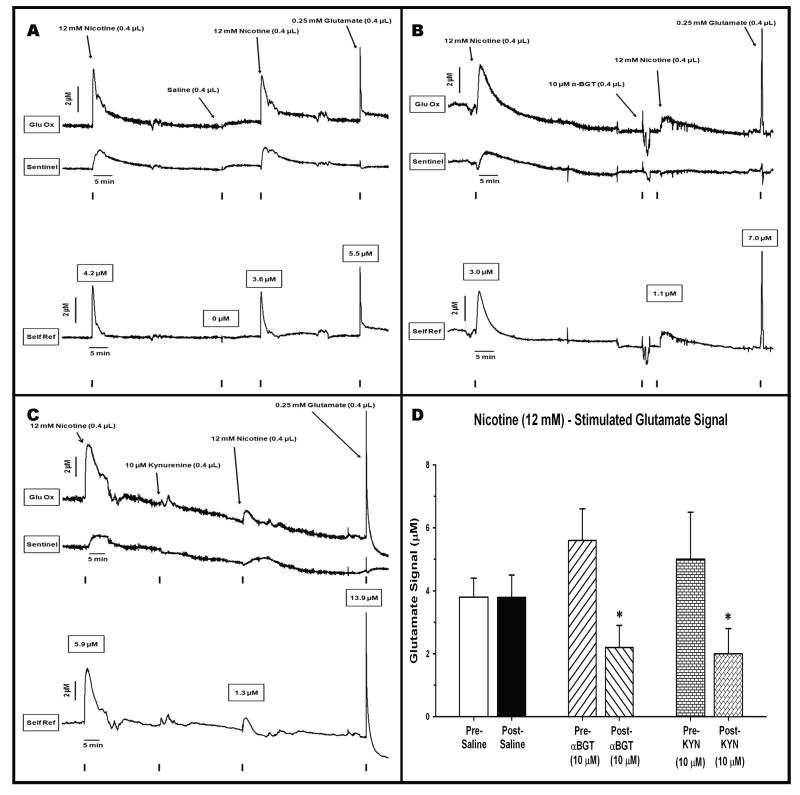
Similar articles
-
Localized infusions of the partial alpha 7 nicotinic receptor agonist SSR180711 evoke rapid and transient increases in prefrontal glutamate release.Neuroscience. 2013;255:55-67. doi: 10.1016/j.neuroscience.2013.09.047. Epub 2013 Oct 1. Neuroscience. 2013. PMID: 24095692
-
Selective potentiation of (α4)3(β2)2 nicotinic acetylcholine receptors augments amplitudes of prefrontal acetylcholine- and nicotine-evoked glutamatergic transients in rats.Biochem Pharmacol. 2013 Nov 15;86(10):1487-96. doi: 10.1016/j.bcp.2013.09.005. Epub 2013 Sep 16. Biochem Pharmacol. 2013. PMID: 24051136 Free PMC article.
-
Presynaptic alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate [3H]d-aspartate release from rat frontal cortex in vitro.Neuropharmacology. 2005 Jul;49(1):59-72. doi: 10.1016/j.neuropharm.2005.01.030. Epub 2005 Apr 21. Neuropharmacology. 2005. PMID: 15992581
-
Glutamatergic contributions to nicotinic acetylcholine receptor agonist-evoked cholinergic transients in the prefrontal cortex.J Neurosci. 2008 Apr 2;28(14):3769-80. doi: 10.1523/JNEUROSCI.5251-07.2008. J Neurosci. 2008. PMID: 18385335 Free PMC article.
-
alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate dopamine release in vitro and in vivo in the rat prefrontal cortex.Eur J Neurosci. 2009 Feb;29(3):539-50. doi: 10.1111/j.1460-9568.2009.06613.x. Epub 2009 Jan 28. Eur J Neurosci. 2009. PMID: 19187266
Cited by
-
Effects of age and acute ethanol on glutamatergic neurotransmission in the medial prefrontal cortex of freely moving rats using enzyme-based microelectrode amperometry.PLoS One. 2015 Apr 30;10(4):e0125567. doi: 10.1371/journal.pone.0125567. eCollection 2015. PLoS One. 2015. PMID: 25927237 Free PMC article.
-
Targeting glutamate homeostasis for potential treatment of nicotine dependence.Brain Res Bull. 2016 Mar;121:1-8. doi: 10.1016/j.brainresbull.2015.11.010. Epub 2015 Nov 14. Brain Res Bull. 2016. PMID: 26589642 Free PMC article. Review.
-
Fluctuations in endogenous kynurenic acid control hippocampal glutamate and memory.Neuropsychopharmacology. 2011 Oct;36(11):2357-67. doi: 10.1038/npp.2011.127. Epub 2011 Jul 27. Neuropsychopharmacology. 2011. PMID: 21796108 Free PMC article.
-
Kynurenic Acid in Schizophrenia: A Systematic Review and Meta-analysis.Schizophr Bull. 2017 Jul 1;43(4):764-777. doi: 10.1093/schbul/sbw221. Schizophr Bull. 2017. PMID: 28187219 Free PMC article.
-
Effects of chronic inhalation of electronic cigarettes containing nicotine on glial glutamate transporters and α-7 nicotinic acetylcholine receptor in female CD-1 mice.Prog Neuropsychopharmacol Biol Psychiatry. 2017 Jul 3;77:1-8. doi: 10.1016/j.pnpbp.2017.03.017. Epub 2017 Mar 27. Prog Neuropsychopharmacol Biol Psychiatry. 2017. PMID: 28347687 Free PMC article.
References
-
- Alkondon M, Pereira E, Cortes WS, Maelicke A, Albuquergue EX. Choline is a selective agonsit of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. European Journal of Neuroscience. 1997;9(12):2734–2742. - PubMed
-
- Alkondon M, Albuquerque E. Subtype-specific inhibition of nicotinic acetylcholine receptors by choline: a regulatory pathway. Journal of Pharmacology and Experimental Therapeutics. 2006;318(1):268–275. - PubMed
-
- Bitner RS, Bunnelle WH, Anderson DJ, Briggs CA, Buccafusco J, Curzon P, Decker MW, Frost JM, Gronlien J, Gubbins E, Li J, Malysz J, Markosyan S, Marsch K, Meyer MD, Nikkel AL, Radek RJ, Robb HM, Timmerman D, Sullivan JP, Gopalakrishnan M. Broad-spectrum efficacy across cognitive domains by α7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. Journal of Neuroscience. 2007;27(39):10578–10587. - PMC - PubMed
-
- Bruno JP, Gash C, Martin B, Zmarowski A, Pomerleau F, Burmeister J, Huettl P, Gerhardt GA. Second-by-second measurement of acetylcholine release in prefrontal cortex. European Journal of Neuroscience. 2006;24(10):2749–2757. - PubMed
-
- Buchanan RW, Conley RR, Dickinson D, Ball MP, Feldman S, Gold JM, McMahon RP. Galantamine for the treatment of cognitive impairments in people with schizophrenia. American Journal of Psychiatry. 2008;165:82–89. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous