

An allosteric mechanism for inhibiting HIV-1 integrase with a small molecule
- PMID: 19638533
- PMCID: PMC2769043
- DOI: 10.1124/mol.109.058883
An allosteric mechanism for inhibiting HIV-1 integrase with a small molecule
Abstract
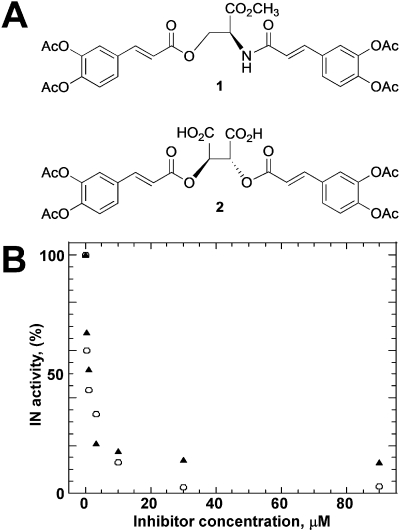
HIV-1 integrase (IN) is a validated target for developing antiretroviral inhibitors. Using affinity acetylation and mass spectrometric (MS) analysis, we previously identified a tetra-acetylated inhibitor (2E)-3-[3,4-bis(acetoxy)phenyl]-2-propenoate-N-[(2E)-3-[3,4-bis(acetyloxy)phenyl]-1-oxo-2-propenyl]-L-serine methyl ester; compound 1] that selectively modified Lys173 at the IN dimer interface. Here we extend our efforts to dissect the mechanism of inhibition and structural features that are important for the selective binding of compound 1. Using a subunit exchange assay, we found that the inhibitor strongly modulates dynamic interactions between IN subunits. Restricting such interactions does not directly interfere with IN binding to DNA substrates or cellular cofactor lens epithelium-derived growth factor, but it compromises the formation of the fully functional nucleoprotein complex. Studies comparing compound 1 with a structurally related IN inhibitor, the tetra-acetylated-chicoric acid derivative (2R,3R)-2,3-bis[[(2E)-3-[3,4-bis(acetyloxy)phenyl]-1-oxo-2-propen-1-yl]oxy]-butanedioic acid (compound 2), indicated striking mechanistic differences between these agents. The structures of the two inhibitors differ only in their central linker regions, with compounds 1 and 2 containing a single methyl ester group and two carboxylic acids, respectively. MS experiments highlighted the importance of these structural differences for selective binding of compound 1 to the IN dimer interface. Moreover, molecular modeling of compound 1 complexed to IN identified a potential inhibitor binding cavity and provided structural clues regarding a possible role of the central methyl ester group in establishing an extensive hydrogen bonding network with both interacting subunits. The proposed mechanism of action and binding site for the small-molecule inhibitor identified in the present study provide an attractive venue for developing allosteric inhibitors of HIV-1 IN.
Figures






References
-
- Al-Mawsawi LQ, Christ F, Dayam R, Debyser Z, Neamati N. (2008) Inhibitory profile of a LEDGF/p75 peptide against HIV-1 integrase: insight into integrase-DNA complex formation and catalysis. FEBS Lett 582:1425–1430 - PubMed
-
- Busschots K, De Rijck J, Christ F, Debyser Z. (2009) In search of small molecules blocking interactions between HIV proteins and intracellular cofactors. Mol Biosyst 5:21–31 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
