

Maximum likelihood genome assembly
- PMID: 19645596
- PMCID: PMC3154397
- DOI: 10.1089/cmb.2009.0047
Maximum likelihood genome assembly
Abstract
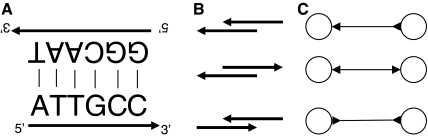
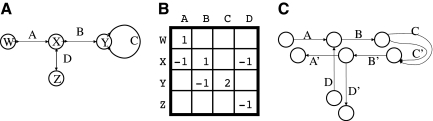
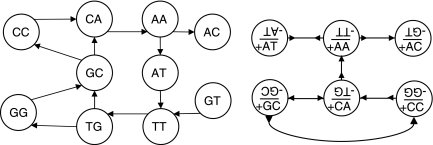
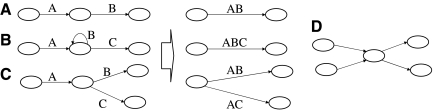
Whole genome shotgun assembly is the process of taking many short sequenced segments (reads) and reconstructing the genome from which they originated. We demonstrate how the technique of bidirected network flow can be used to explicitly model the double-stranded nature of DNA for genome assembly. By combining an algorithm for the Chinese Postman Problem on bidirected graphs with the construction of a bidirected de Bruijn graph, we are able to find the shortest double-stranded DNA sequence that contains a given set of k-long DNA molecules. This is the first exact polynomial time algorithm for the assembly of a double-stranded genome. Furthermore, we propose a maximum likelihood framework for assembling the genome that is the most likely source of the reads, in lieu of the standard maximum parsimony approach (which finds the shortest genome subject to some constraints). In this setting, we give a bidirected network flow-based algorithm that, by taking advantage of high coverage, accurately estimates the copy counts of repeats in a genome. Our second algorithm combines these predicted copy counts with matepair data in order to assemble the reads into contigs. We run our algorithms on simulated read data from Escherichia coli and predict copy counts with extremely high accuracy, while assembling long contigs.
Figures
References
-
- Ahuja R.K. Magnanti T.L. Orlin J.B. Network flows: theory, algorithms, and applications. Prentice-Hall; Upper Saddle River, NJ: 1993.
-
- Appa G. Kotnyek B. A bidirected generalization of network matrices. Networks. 2006;47:185–198.
-
- Chaisson M. Pevzner P.A. Tang H. Fragment assembly with short reads. Bioinformatics. 2004;20:2067–2074. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
