

Identification of a physiologically relevant endogenous ligand for PPARalpha in liver
- PMID: 19646743
- PMCID: PMC2725194
- DOI: 10.1016/j.cell.2009.05.036
Identification of a physiologically relevant endogenous ligand for PPARalpha in liver
Abstract
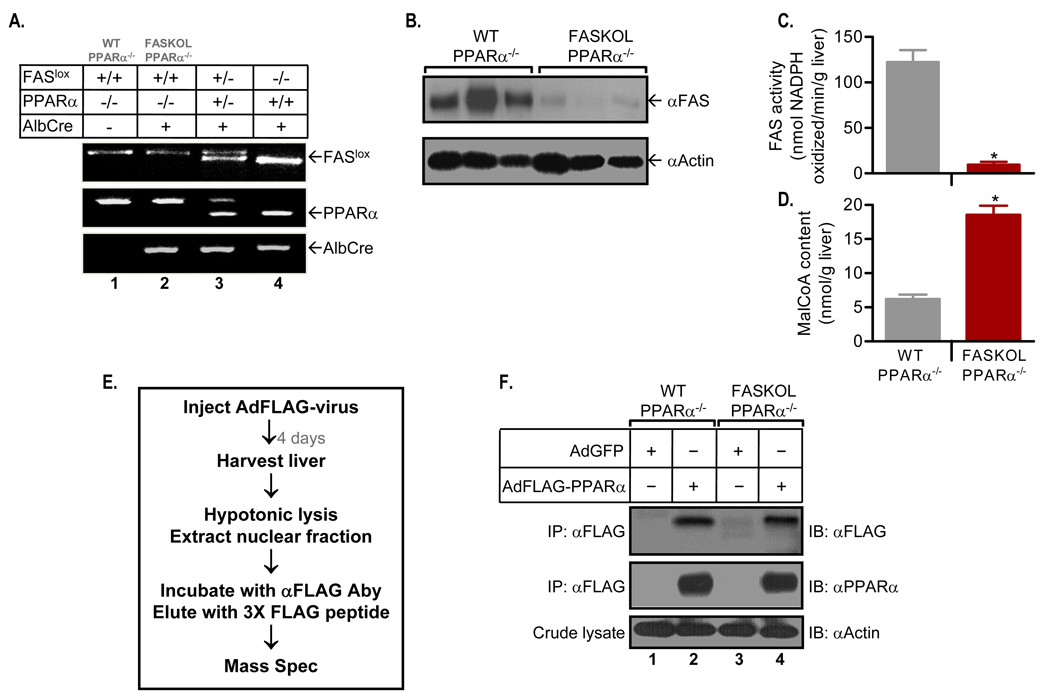
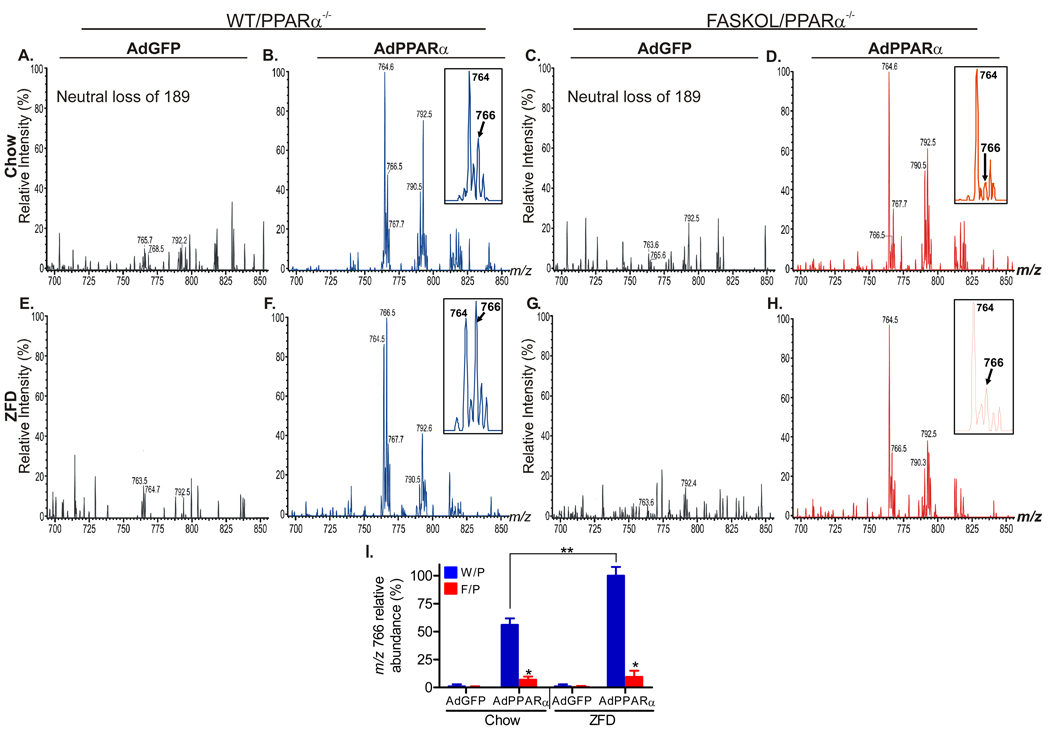
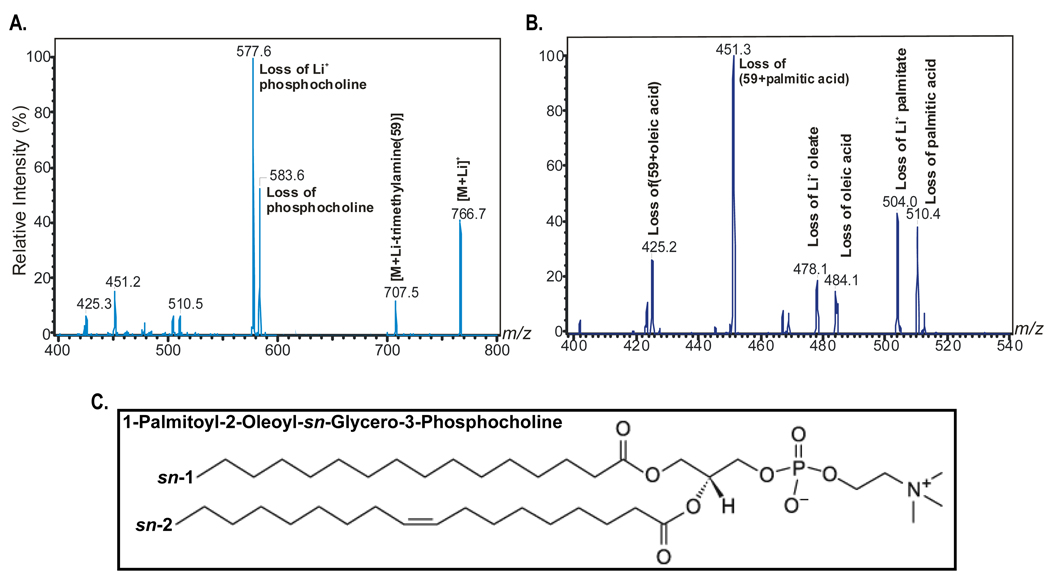
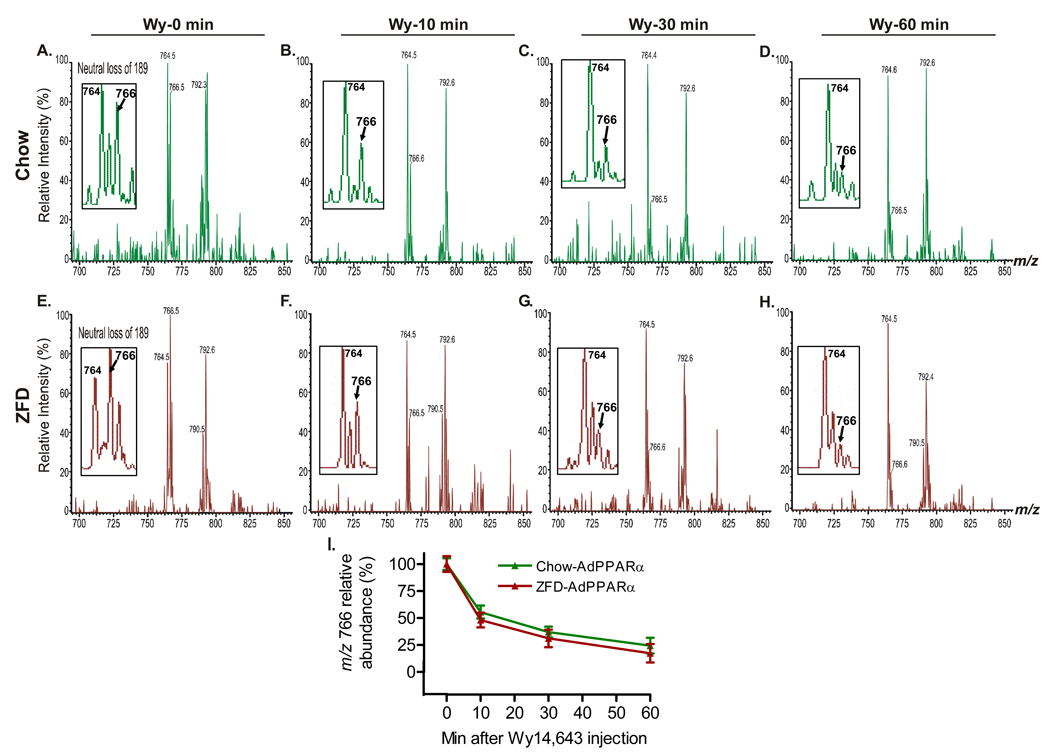
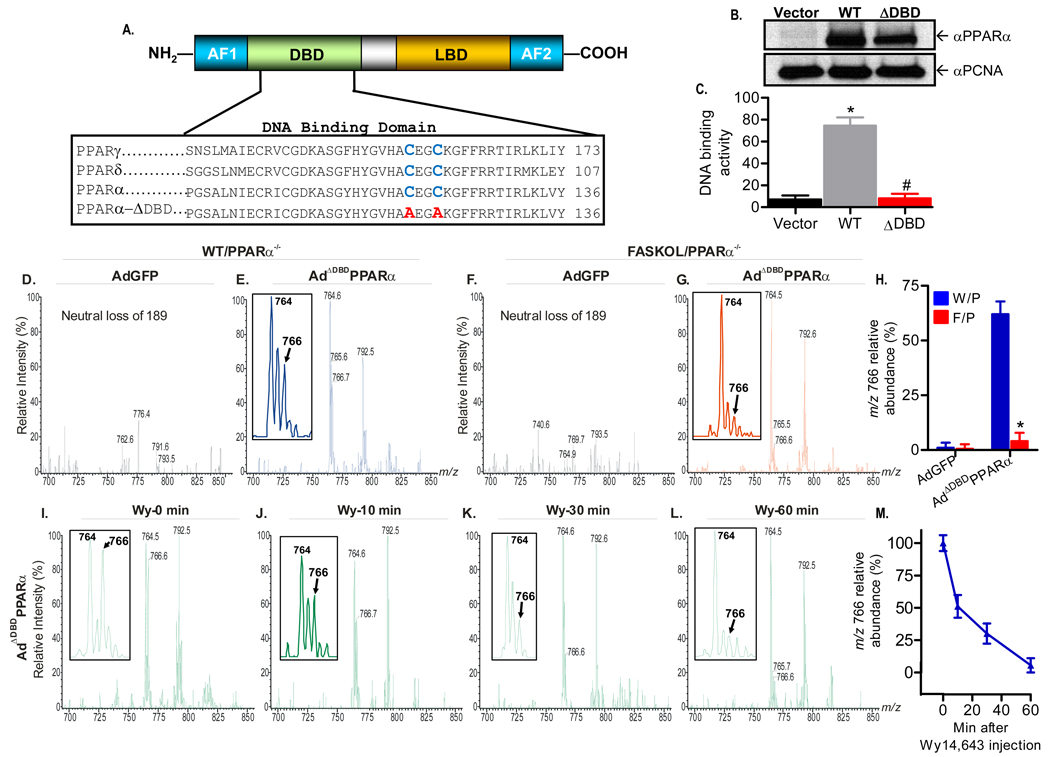
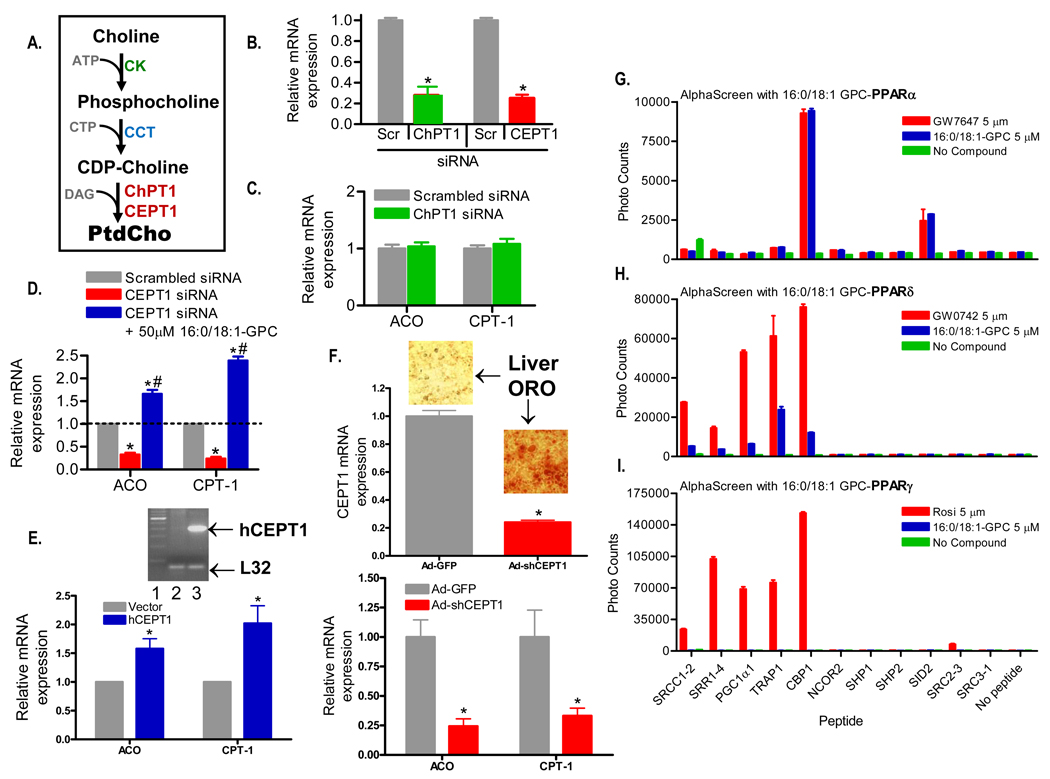
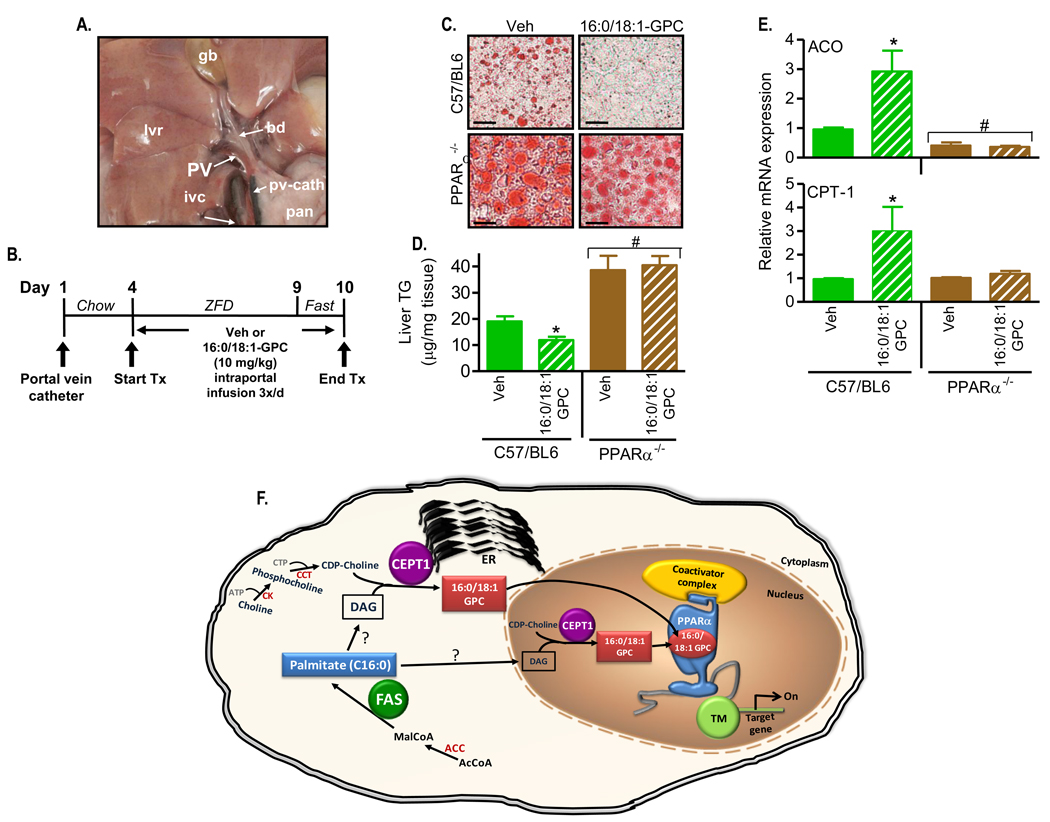
The nuclear receptor PPARalpha is activated by drugs to treat human disorders of lipid metabolism. Its endogenous ligand is unknown. PPARalpha-dependent gene expression is impaired with inactivation of fatty acid synthase (FAS), suggesting that FAS is involved in generation of a PPARalpha ligand. Here we demonstrate the FAS-dependent presence of a phospholipid bound to PPARalpha isolated from mouse liver. Binding was increased under conditions that induce FAS activity and displaced by systemic injection of a PPARalpha agonist. Mass spectrometry identified the species as 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-GPC). Knockdown of Cept1, required for phosphatidylcholine synthesis, suppressed PPARalpha-dependent gene expression. Interaction of 16:0/18:1-GPC with the PPARalpha ligand-binding domain and coactivator peptide motifs was comparable to PPARalpha agonists, but interactions with PPARdelta were weak and none were detected with PPARgamma. Portal vein infusion of 16:0/18:1-GPC induced PPARalpha-dependent gene expression and decreased hepatic steatosis. These data suggest that 16:0/18:1-GPC is a physiologically relevant endogenous PPARalpha ligand.
Figures
References
-
- Ayers SD, Nedrow KL, Gillilan RE, Noy N. Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPARgamma by FABP4. Biochemistry. 2007;46:6744–6752. - PubMed
-
- Barter PJ, Rye KA. Is there a role for fibrates in the management of dyslipidemia in the metabolic syndrome? Arterioscler Thromb Vasc Biol. 2008;28:39–46. - PubMed
-
- Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. - PubMed
-
- Bernal-Mizrachi C, Weng S, Feng C, Finck BN, Knutsen RH, Leone TC, Coleman T, Mecham RP, Kelly DP, Semenkovich CF. Dexamethasone induction of hypertension and diabetes is PPARalpha-dependent in LDL receptor-null mice. Nat Med. 2003;9:1069–1075. - PubMed
-
- Billas IM, Moulinier L, Rochel N, Moras D. Crystal structure of the ligand-binding domain of the ultraspiracle protein USP, the ortholog of retinoid X receptors in insects. J Biol Chem. 2001;276:7465–7474. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R37 DK034388/DK/NIDDK NIH HHS/United States
- DK076729/DK/NIDDK NIH HHS/United States
- HL089301/HL/NHLBI NIH HHS/United States
- DK20579/DK/NIDDK NIH HHS/United States
- P30 DK056341/DK/NIDDK NIH HHS/United States
- P60 DK020579/DK/NIDDK NIH HHS/United States
- P41 RR00954/RR/NCRR NIH HHS/United States
- P41 RR000954/RR/NCRR NIH HHS/United States
- P50 HL083762/HL/NHLBI NIH HHS/United States
- DK56341/DK/NIDDK NIH HHS/United States
- R01 HL089301/HL/NHLBI NIH HHS/United States
- R01 DK076729/DK/NIDDK NIH HHS/United States
- R37 DK34388/DK/NIDDK NIH HHS/United States
- P30 DK020579/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
