

Computational design of affinity and specificity at protein-protein interfaces
- PMID: 19646858
- PMCID: PMC2882636
- DOI: 10.1016/j.sbi.2009.07.005
Computational design of affinity and specificity at protein-protein interfaces
Abstract
The computer-based design of protein-protein interactions is a rigorous test of our understanding of molecular recognition and an attractive approach for creating novel tools for cell and molecular research. Considerable attention has been placed on redesigning the affinity and specificity of naturally occurring interactions. Several studies have shown that reducing the desolvation costs for binding while preserving shape complimentarity and hydrogen bonding is an effective strategy for improving binding affinities. In favorable cases specificity has been designed by focusing only on interactions with the target protein, while in cases with closely related off-target proteins it has been necessary to explicitly disfavor unwanted binding partners. The rational design of protein-protein interactions from scratch is still an unsolved problem, but recent developments in flexible backbone design and energy functions hold promise for the future.
Figures
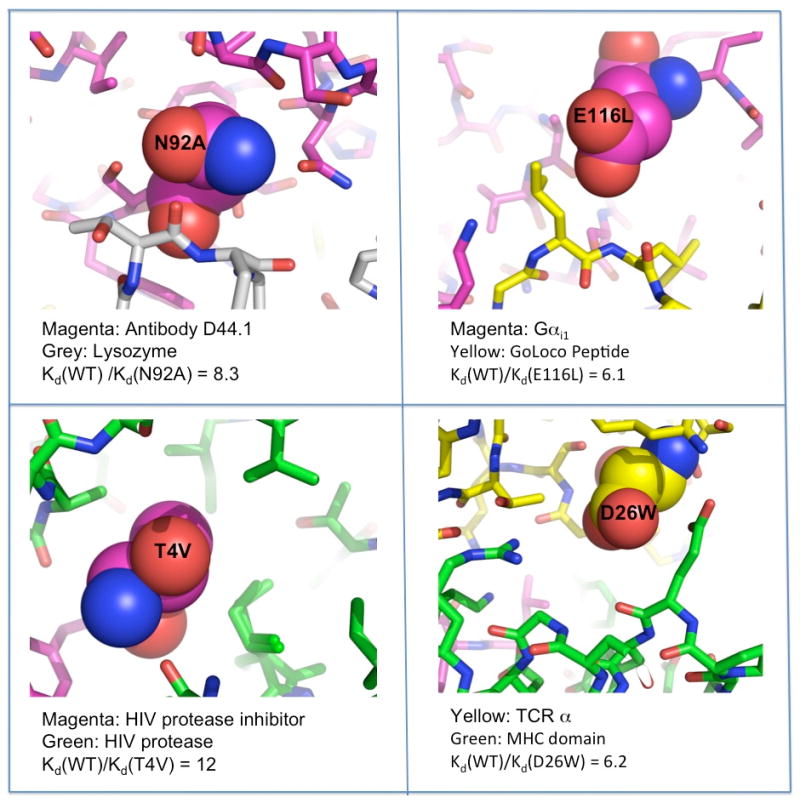
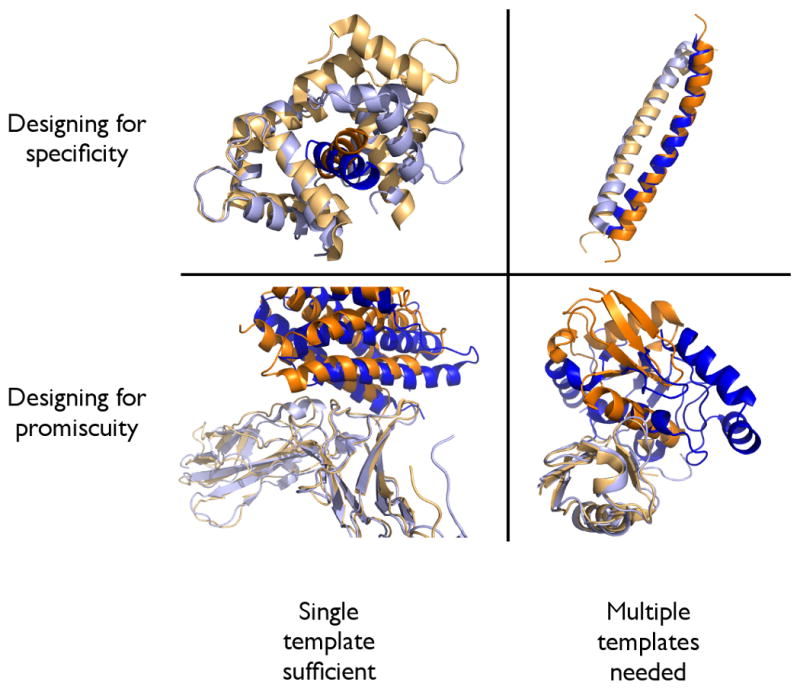
References
-
- Boas FE, Harbury PB. Potential energy functions for protein design. Curr Opin Struct Biol. 2007;17:199–204. - PubMed
-
- Cohen M, Reichmann D, Neuvirth H, Schreiber G. Similar chemistry, but different bond preferences in inter versus intra-protein interactions. Proteins. 2008;72:741–753. - PubMed
-
- Ofran Y, Rost B. Analysing six types of protein-protein interfaces. J Mol Biol. 2003;325:377–387. - PubMed
-
- Reichmann D, Phillip Y, Carmi A, Schreiber G. On the contribution of water-mediated interactions to protein-complex stability. Biochemistry. 2008;47:1051–1060. - PubMed
Commented References
-
- Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–1176. - PMC - PubMed
-
Affinity enhancing mutations were identified by searching for mutations that increase the favorability of electrostatic solvation and interaction energies calculated with the Poisson-Boltzmann equation.
-
- Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. - PMC - PubMed
-
This is the first large-scale demonstration of computational protein design being used to redesign binding specificities. Explicit consideration of off-target binders was required to achieve specificity.
-
- Humphris EL, Kortemme T. Design of multi-specificity in protein interfaces. PLoS Comput Biol. 2007;3:e164. - PMC - PubMed
-
Introduction of multi-constraint design methodology allowed the authors to estimate the degree of “compromise” encoded in sequences of promiscuous proteins. Surprisingly, little evidence for compromise was identified in most cases.
-
- Yosef E, Politi R, Choi MH, Shifman JM. Computational design of calmodulin mutants with up to 900-fold increase in binding specificity. J Mol Biol. 2009;385:1470–1480. - PubMed
-
In this case specificity was achieved by focusing only on the target peptide. Significant structural differences between the target and competing peptides may be the reason explicit negative design was not required.
-
- Humphris EL, Kortemme T. Prediction of Protein-Protein Interface Sequence Diversity Using Flexible Backbone Computational Protein Design. Structure. 2008;16:1777–1788. - PubMed
-
Introducing small perturbations to Cα-Cβ bond vectors allowed for better recapitulation of sequences known to bind the target peptide. This may be an effective strategy for creating directed libraries for protein interface design.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
