

Sensitive and accurate detection of copy number variants using read depth of coverage
- PMID: 19657104
- PMCID: PMC2752127
- DOI: 10.1101/gr.092981.109
Sensitive and accurate detection of copy number variants using read depth of coverage
Abstract
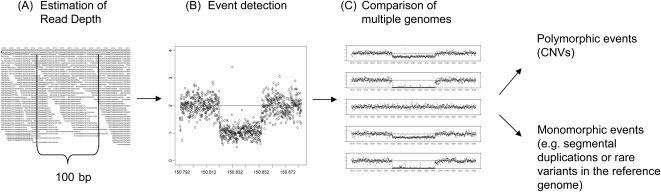
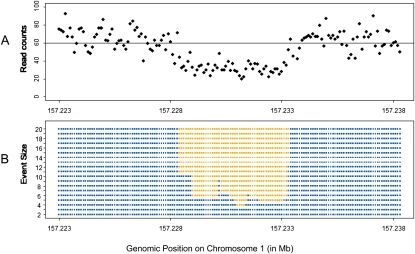
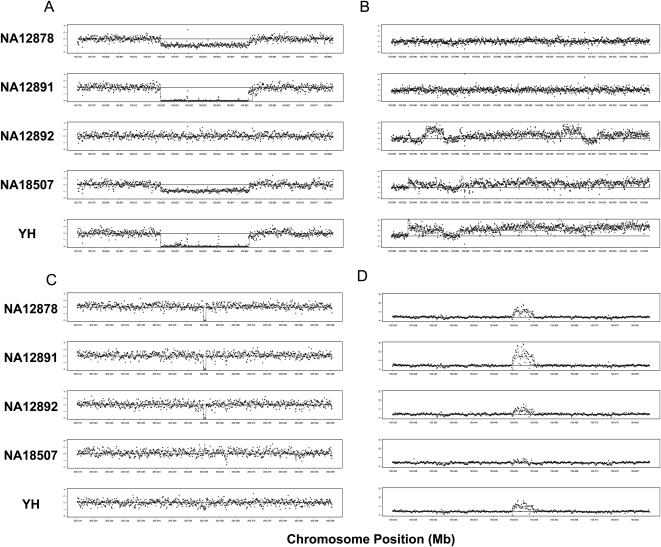
Methods for the direct detection of copy number variation (CNV) genome-wide have become effective instruments for identifying genetic risk factors for disease. The application of next-generation sequencing platforms to genetic studies promises to improve sensitivity to detect CNVs as well as inversions, indels, and SNPs. New computational approaches are needed to systematically detect these variants from genome sequence data. Existing sequence-based approaches for CNV detection are primarily based on paired-end read mapping (PEM) as reported previously by Tuzun et al. and Korbel et al. Due to limitations of the PEM approach, some classes of CNVs are difficult to ascertain, including large insertions and variants located within complex genomic regions. To overcome these limitations, we developed a method for CNV detection using read depth of coverage. Event-wise testing (EWT) is a method based on significance testing. In contrast to standard segmentation algorithms that typically operate by performing likelihood evaluation for every point in the genome, EWT works on intervals of data points, rapidly searching for specific classes of events. Overall false-positive rate is controlled by testing the significance of each possible event and adjusting for multiple testing. Deletions and duplications detected in an individual genome by EWT are examined across multiple genomes to identify polymorphism between individuals. We estimated error rates using simulations based on real data, and we applied EWT to the analysis of chromosome 1 from paired-end shotgun sequence data (30x) on five individuals. Our results suggest that analysis of read depth is an effective approach for the detection of CNVs, and it captures structural variants that are refractory to established PEM-based methods.
Figures
References
-
- Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum Mol Genet. 2003;12(Spec. no. 2):R145–R152. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials