

Modeling sequence evolution in acute HIV-1 infection
- PMID: 19660475
- PMCID: PMC2760689
- DOI: 10.1016/j.jtbi.2009.07.038
Modeling sequence evolution in acute HIV-1 infection
Erratum in
- J Theor Biol. 2012 Mar 21;297:187
Abstract
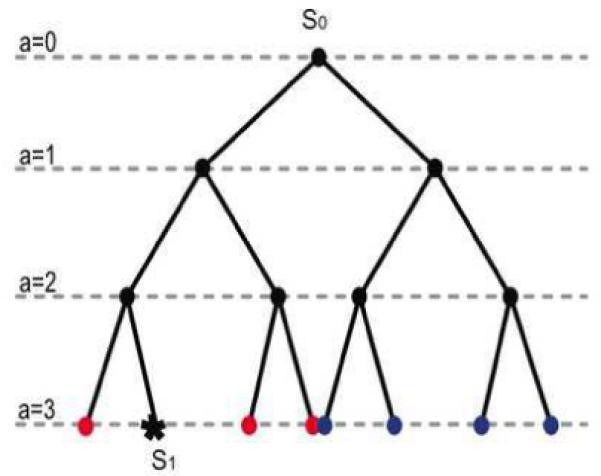
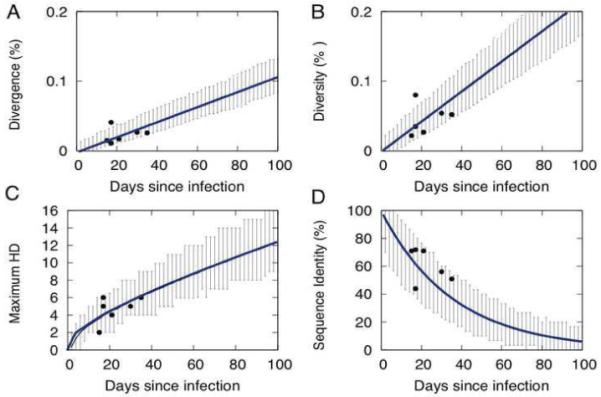
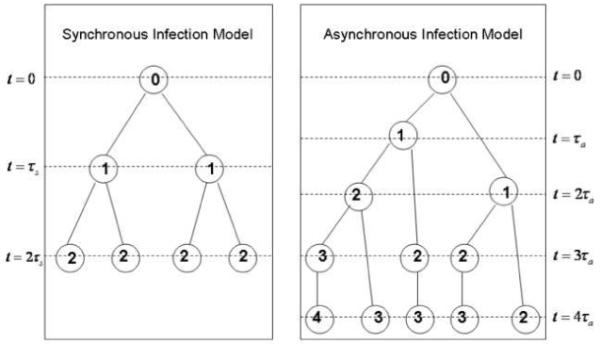
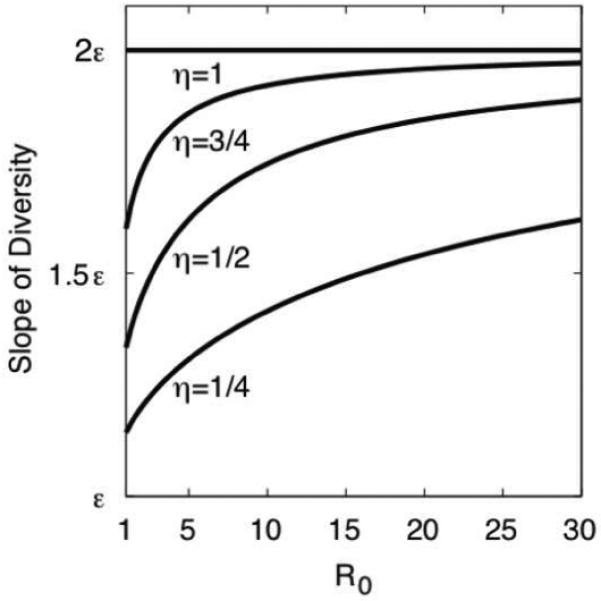
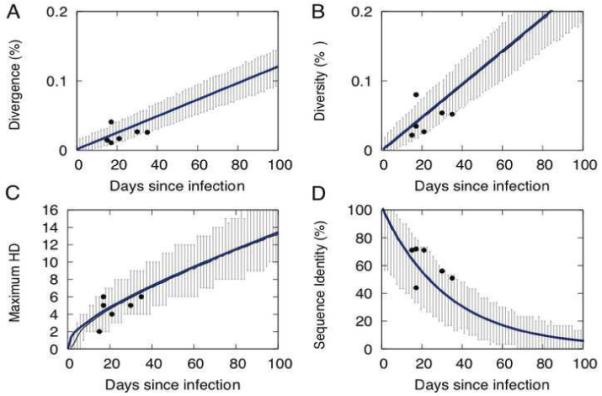
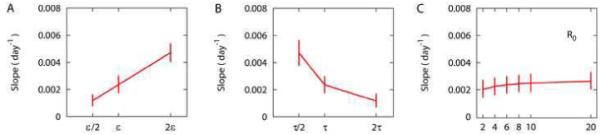
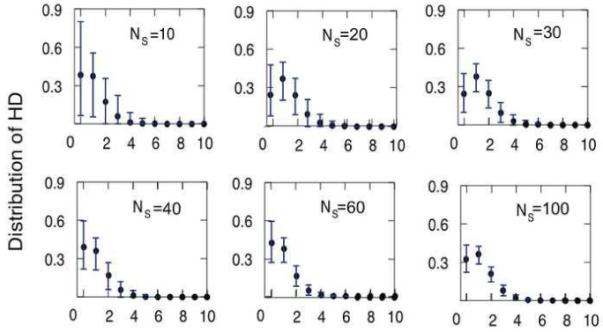
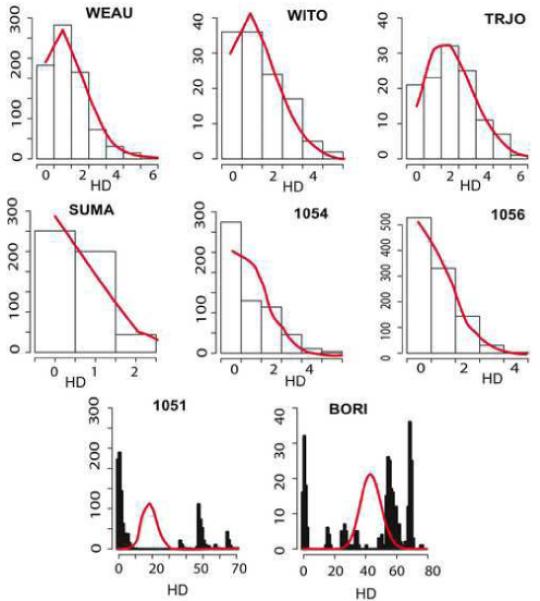
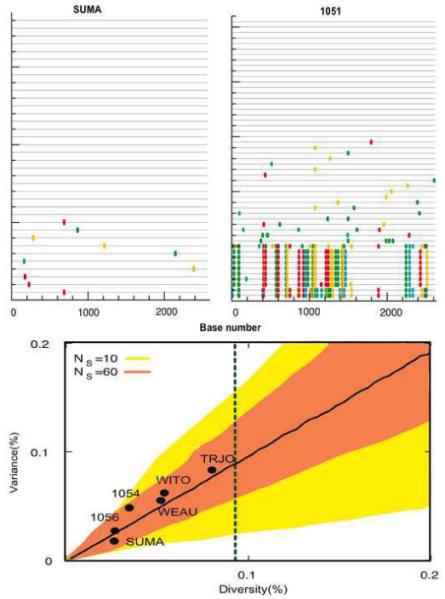
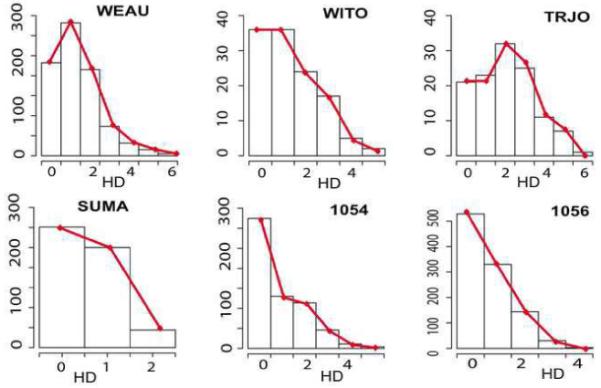
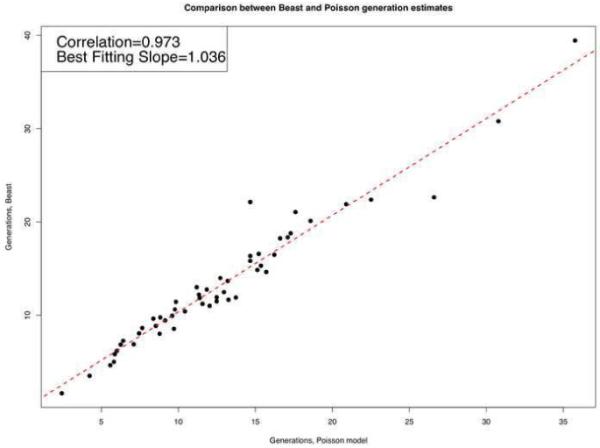
We describe a mathematical model and Monte Carlo (MC) simulation of viral evolution during acute infection. We consider both synchronous and asynchronous processes of viral infection of new target cells. The model enables an assessment of the expected sequence diversity in new HIV-1 infections originating from a single transmitted viral strain, estimation of the most recent common ancestor (MRCA) of the transmitted viral lineage, and estimation of the time to coalesce back to the MRCA. We also calculate the probability of the MRCA being the transmitted virus or an evolved variant. Excluding insertions and deletions, we assume HIV-1 evolves by base substitution without selection pressure during the earliest phase of HIV-1 infection prior to the immune response. Unlike phylogenetic methods that follow a lineage backwards to coalescence, we compare the observed data to a model of the diversification of a viral population forward in time. To illustrate the application of these methods, we provide detailed comparisons of the model and simulations results to 306 envelope sequences obtained from eight newly infected subjects at a single time point. The data from 68 patients were in good agreement with model predictions, and hence compatible with a single-strain infection evolving under no selection pressure. The diversity of the samples from the other two patients was too great to be explained by the model, suggesting multiple HIV-1-strains were transmitted. The model can also be applied to longitudinal patient data to estimate within-host viral evolutionary parameters.
Figures
References
-
- Abrahams MR, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, Salazar-Gonzalez JF, Bhattacharya T, Chu H, Hoffman I, Galvin S, Mapanje C, Kazembe P, Thebus R, Fiscus S, Hide W, Cohen MS, Karim SA, Haynes BF, Shaw GM, Hahn BH, Korber BT, Swanstrom R, Williamson C. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J Virol. 2009;83:3556–67. - PMC - PubMed
-
- Achaz G, Palmer S, Kearney M, Maldarelli F, Mellors JW, Coffin JM, Wakeley J. A robust measure of HIV-1 population turnover within chronically infected individuals. Mol Biol Evol. 2004;21:1902–12. - PubMed
-
- Casella G, Berger RL. Statistical inference. Brooks/Cole Pub. Co.; Pacific Grove, Calif.: 1990.
Publication types
MeSH terms
Grants and funding
- P30-AI078498/AI/NIAID NIH HHS/United States
- U01 AI067854/AI/NIAID NIH HHS/United States
- RR06555/RR/NCRR NIH HHS/United States
- R37 AI028433/AI/NIAID NIH HHS/United States
- AI67854/AI/NIAID NIH HHS/United States
- R01 AI028433/AI/NIAID NIH HHS/United States
- R01 RR006555/RR/NCRR NIH HHS/United States
- U19 AI067854/AI/NIAID NIH HHS/United States
- AI27767/AI/NIAID NIH HHS/United States
- P30 AI027767/AI/NIAID NIH HHS/United States
- R01 OD011095/OD/NIH HHS/United States
- AI028433/AI/NIAID NIH HHS/United States
- AI083115/AI/NIAID NIH HHS/United States
- R01 AI083115/AI/NIAID NIH HHS/United States
- P30 AI078498/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
