

A method for helical RNA global structure determination in solution using small-angle x-ray scattering and NMR measurements
- PMID: 19666030
- PMCID: PMC2760655
- DOI: 10.1016/j.jmb.2009.08.001
A method for helical RNA global structure determination in solution using small-angle x-ray scattering and NMR measurements
Abstract
We report a "top-down" method that uses mainly duplexes' global orientations and overall molecular dimension and shape restraints, which were extracted from experimental NMR and small-angle X-ray scattering data, respectively, to determine global architectures of RNA molecules consisting of mostly A-form-like duplexes. The method is implemented in the G2G (from global measurement to global structure) toolkit of programs. We demonstrate the efficiency and accuracy of the method by determining the global structure of a 71-nt RNA using experimental data. The backbone root-mean-square deviation of the ensemble of the calculated global structures relative to the X-ray crystal structure is 3.0+/-0.3 A using the experimental data and is only 2.5+/-0.2 A for the three duplexes that were orientation restrained during the calculation. The global structure simplifies interpretation of multidimensional nuclear Overhauser spectra for high-resolution structure determination. The potential general application of the method for RNA structure determination is discussed.
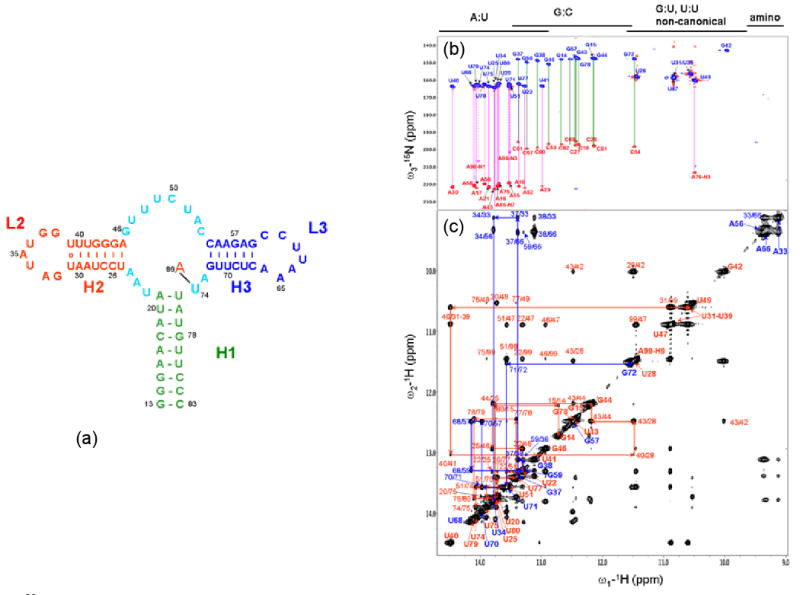
Figures











References
-
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. - PubMed
-
- Tucker BJ, Breaker RR. Riboswitches as versatile gene control elements. Curr Opin Struct Biol. 2005;15:342–348. - PubMed
-
- Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–157. - PubMed
-
- Blackburn EH. Telomerases. Annu Rev Biochem. 1992;61:113–129. - PubMed
-
- Moore PB, Steitz TA. The involvement of RNA in ribosome function. Nature. 2002;418:229–235. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
