

Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis
- PMID: 19666474
- PMCID: PMC2785637
- DOI: 10.1074/jbc.M109.031336
Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis
Abstract
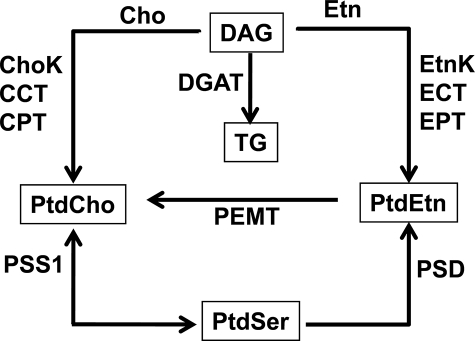
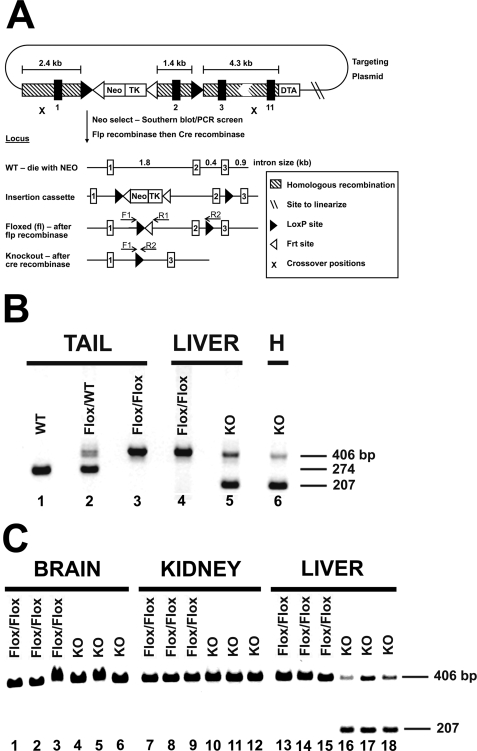
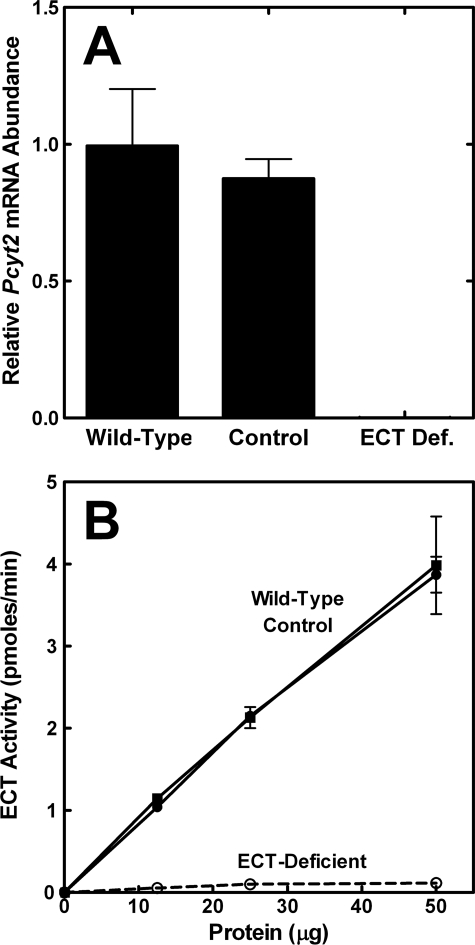
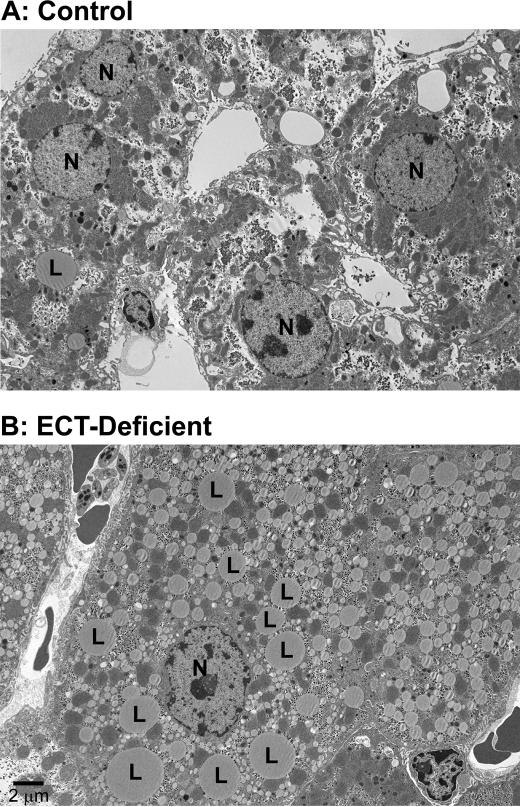
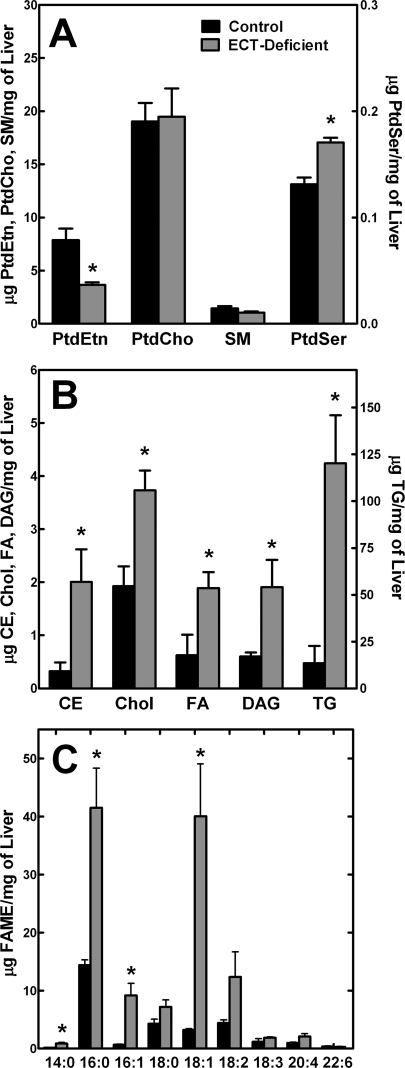
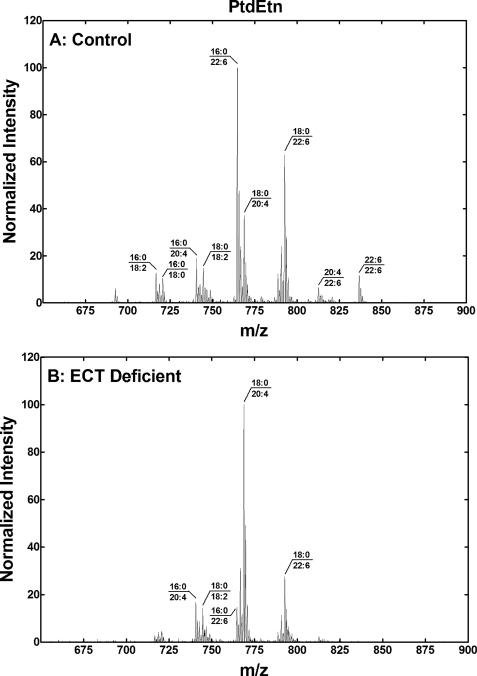
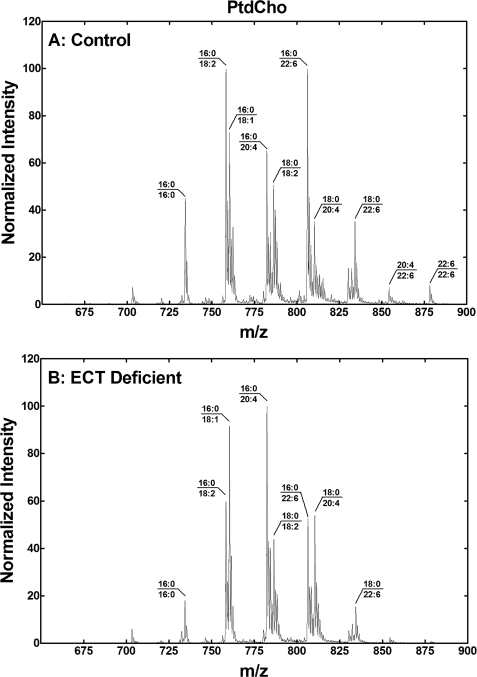
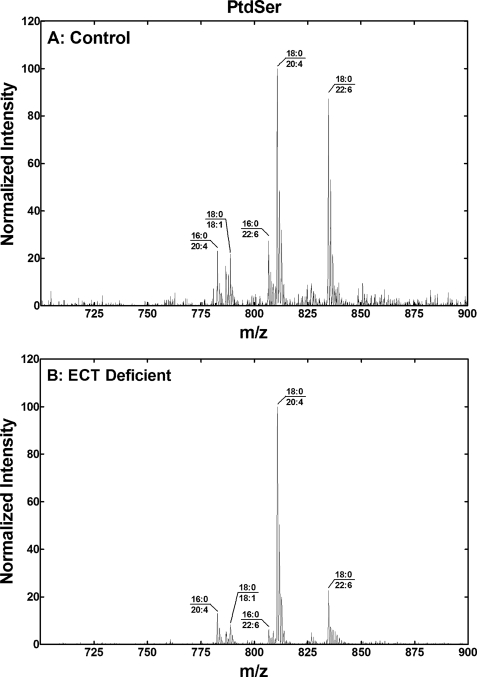
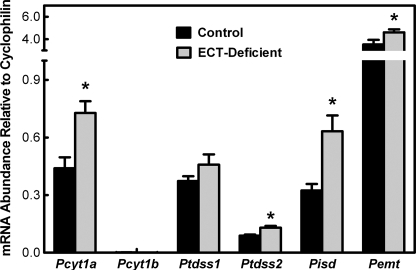
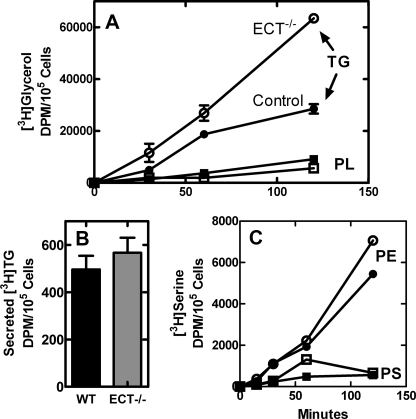
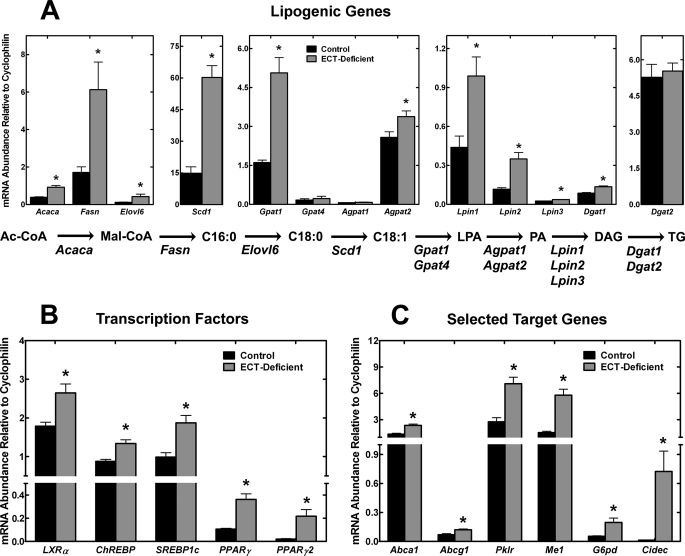
Phosphoethanolamine cytidylyltransferase (ECT) catalyzes the rate-controlling step in a major pathway for the synthesis of phosphatidylethanolamine (PtdEtn). Hepatocyte-specific deletion of the ECT gene in mice resulted in normal appearing animals without overt signs of liver injury or inflammation. The molecular species of PtdEtn in the ECT-deficient livers were significantly altered compared with controls and matched the composition of the phosphatidylserine (PtdSer) pool, illustrating the complete reliance on the PtdSer decarboxylase pathway for PtdEtn synthesis. PtdSer structure was controlled by the substrate specificity of PtdSer synthase that selectively converted phosphatidylcholine molecular species containing stearate paired with a polyunsaturated fatty acid to PtdSer. There was no evidence for fatty acid remodeling of PtdEtn. The elimination of diacylglycerol utilization by the CDP-ethanolamine pathway led to a 10-fold increase in triacylglycerols in the ECT-deficient hepatocytes that became engorged with lipid droplets. Triacylglycerol accumulation was associated with a significant elevation in the expression of the transcription factors and target genes that drive de novo lipogenesis. The absence of the ECT pathway for diacylglycerol utilization at the endoplasmic reticulum triggers increased fatty acid synthesis to support the formation of triacylglycerols leading to liver steatosis.
Figures
Similar articles
-
Cloning and expression of mouse liver phosphatidylserine synthase-1 cDNA. Overexpression in rat hepatoma cells inhibits the CDP-ethanolamine pathway for phosphatidylethanolamine biosynthesis.J Biol Chem. 1998 Mar 27;273(13):7293-302. doi: 10.1074/jbc.273.13.7293. J Biol Chem. 1998. PMID: 9516423
-
The CDP-ethanolamine pathway and phosphatidylserine decarboxylation generate different phosphatidylethanolamine molecular species.J Biol Chem. 2007 Sep 28;282(39):28362-28372. doi: 10.1074/jbc.M703786200. Epub 2007 Aug 2. J Biol Chem. 2007. PMID: 17673461
-
The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice.J Biol Chem. 2009 Sep 18;284(38):25704-13. doi: 10.1074/jbc.M109.023846. Epub 2009 Jul 22. J Biol Chem. 2009. PMID: 19625253 Free PMC article.
-
Metabolic and molecular aspects of ethanolamine phospholipid biosynthesis: the role of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2).Biochem Cell Biol. 2007 Jun;85(3):283-300. doi: 10.1139/o07-006. Biochem Cell Biol. 2007. PMID: 17612623 Review.
-
Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells.Biochim Biophys Acta. 2013 Mar;1831(3):543-54. doi: 10.1016/j.bbalip.2012.08.016. Epub 2012 Aug 29. Biochim Biophys Acta. 2013. PMID: 22960354 Review.
Cited by
-
Slightly different metabolomic profiles are associated with high or low weight duck foie gras.PLoS One. 2022 Jun 28;17(6):e0255707. doi: 10.1371/journal.pone.0255707. eCollection 2022. PLoS One. 2022. PMID: 35763459 Free PMC article.
-
Historical perspective: phosphatidylserine and phosphatidylethanolamine from the 1800s to the present.J Lipid Res. 2018 Jun;59(6):923-944. doi: 10.1194/jlr.R084004. Epub 2018 Apr 16. J Lipid Res. 2018. PMID: 29661786 Free PMC article.
-
Choline transport links macrophage phospholipid metabolism and inflammation.J Biol Chem. 2018 Jul 20;293(29):11600-11611. doi: 10.1074/jbc.RA118.003180. Epub 2018 Jun 7. J Biol Chem. 2018. PMID: 29880645 Free PMC article.
-
Ethanolaminephosphate cytidyltransferase is essential for survival, lipid homeostasis and stress tolerance in Leishmania major.bioRxiv [Preprint]. 2023 Jan 11:2023.01.10.523530. doi: 10.1101/2023.01.10.523530. bioRxiv. 2023. Update in: PLoS Pathog. 2023 Jul 28;19(7):e1011112. doi: 10.1371/journal.ppat.1011112. PMID: 36712124 Free PMC article. Updated. Preprint.
-
Deficiency of phosphatidylethanolamine synthesis: consequences for skeletal muscle.Function (Oxf). 2023 Sep 1;4(6):zqad044. doi: 10.1093/function/zqad044. eCollection 2023. Function (Oxf). 2023. PMID: 37772311 Free PMC article. No abstract available.
References
-
- Bakovic M., Fullerton M. D., Michel V. (2007) Biochem. Cell Biol. 85, 283–300 - PubMed
-
- Vance J. E. (2008) J. Lipid Res. 49, 1377–1387 - PubMed
-
- Nagan N., Zoeller R. A. (2001) Prog. Lipid Res. 40, 199–229 - PubMed
-
- Gorgas K., Teigler A., Komljenovic D., Just W. W. (2006) Biochim. Biophys. Acta 1763, 1511–1526 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
