

GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling
- PMID: 19666519
- PMCID: PMC2728998
- DOI: 10.1073/pnas.0904744106
GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling
Abstract
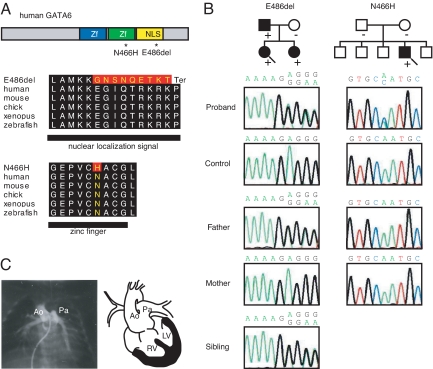
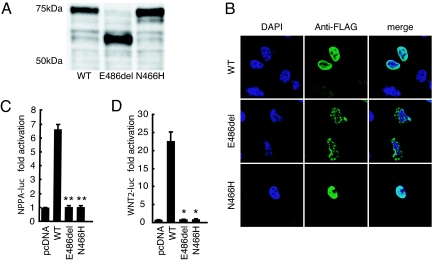
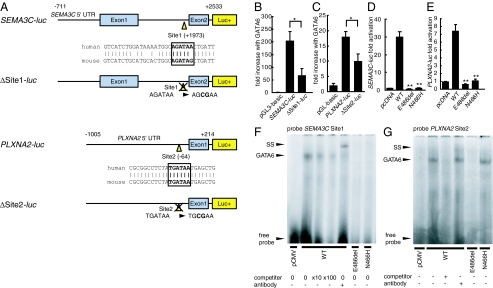
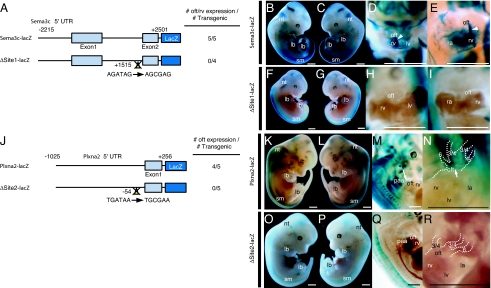
Congenital heart diseases (CHD) occur in nearly 1% of all live births and are the major cause of infant mortality and morbidity. Although an improved understanding of the genetic causes of CHD would provide insight into the underlying pathobiology, the genetic etiology of most CHD remains unknown. Here we show that mutations in the gene encoding the transcription factor GATA6 cause CHD characteristic of a severe form of cardiac outflow tract (OFT) defect, namely persistent truncus arteriosus (PTA). Two different GATA6 mutations were identified by systematic genetic analysis using DNA from patients with PTA. Genes encoding the neurovascular guiding molecule semaphorin 3C (SEMA3C) and its receptor plexin A2 (PLXNA2) appear to be regulated directly by GATA6, and both GATA6 mutant proteins failed to transactivate these genes. Transgenic analysis further suggests that, in the developing heart, the expression of SEMA3C in the OFT/subpulmonary myocardium and PLXNA2 in the cardiac neural crest contributing to the OFT is dependent on GATA transcription factors. Together, our data implicate mutations in GATA6 as genetic causes of CHD involving OFT development, as a result of the disruption of the direct regulation of semaphorin-plexin signaling.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. - PubMed
-
- Thom T, et al. Heart disease and stroke statistics–2006 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;14:e85–151. - PubMed
-
- Williams JM, et al. Factors associated with outcomes of persistent truncus arteriosus. J Am Coll Cardiol. 1999;34:545–553. - PubMed
-
- Srivastava D. Making or breaking the heart: From lineage determination to morphogenesis. Cell. 2006;126:1037–1048. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
