

Proteome-wide prediction of acetylation substrates
- PMID: 19666589
- PMCID: PMC2728972
- DOI: 10.1073/pnas.0906801106
Proteome-wide prediction of acetylation substrates
Abstract
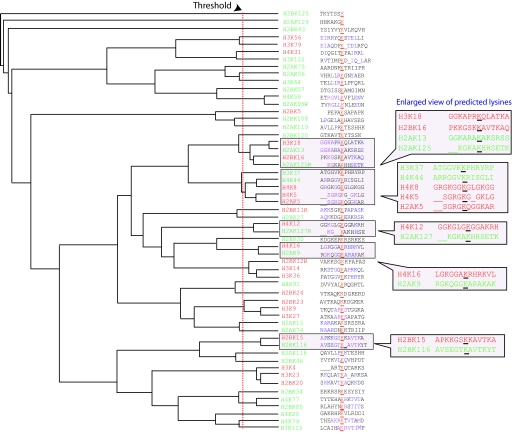
Acetylation is a well-studied posttranslational modification that has been associated with a broad spectrum of biological processes, notably gene regulation. Many studies have contributed to our knowledge of the enzymology underlying acetylation, including efforts to understand the molecular mechanism of substrate recognition by several acetyltransferases, but traditional experiments to determine intrinsic features of substrate site specificity have proven challenging. Here, we combine experimental methods with clustering analysis of protein sequences to predict protein acetylation based on the sequence characteristics of acetylated lysines within histones with our unique prediction tool PredMod. We define a local amino acid sequence composition that represents potential acetylation sites by implementing a clustering analysis of histone and nonhistone sequences. We show that this sequence composition has predictive power on 2 independent experimental datasets of acetylation marks. Finally, we detect acetylation for selected putative substrates using mass spectrometry, and report several nonhistone acetylated substrates in budding yeast. Our approach, combined with more traditional experimental methods, may be useful for identifying acetylated substrates proteome-wide.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
