

Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies
- PMID: 19668216
- PMCID: PMC2746682
- DOI: 10.1038/ng.423
Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies
Abstract
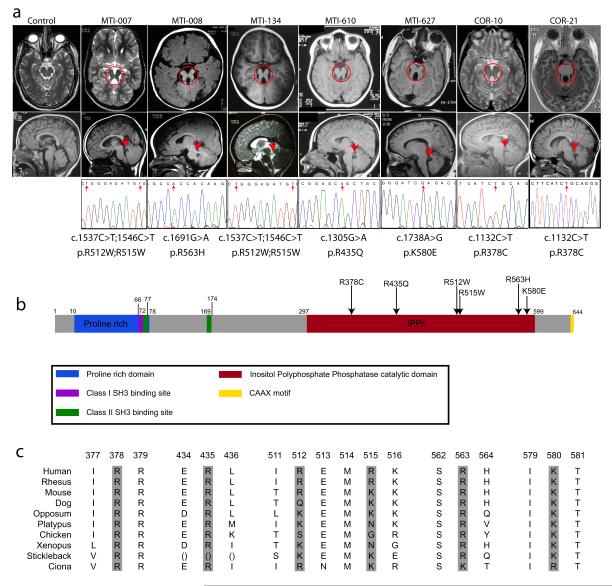
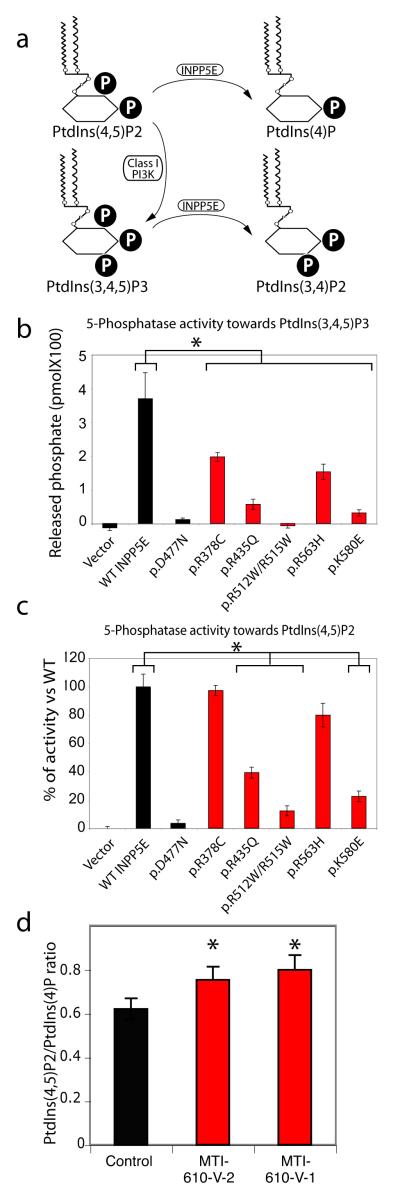
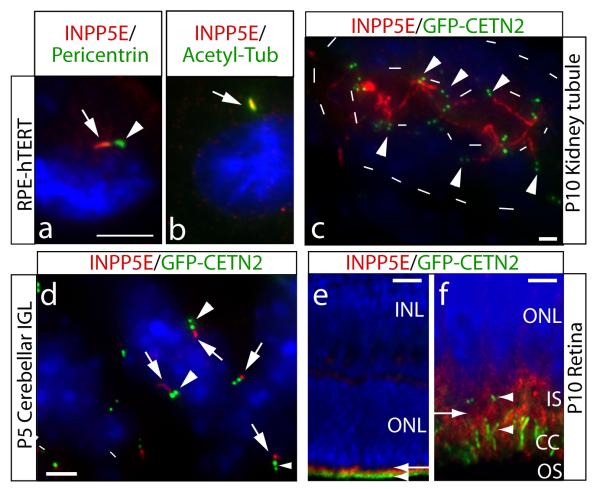
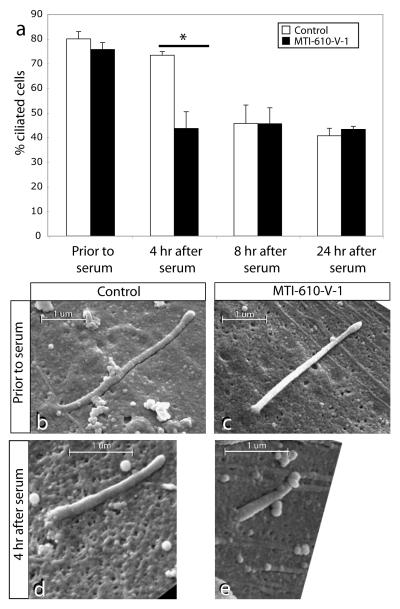
Phosphotidylinositol (PtdIns) signaling is tightly regulated both spatially and temporally by subcellularly localized PtdIns kinases and phosphatases that dynamically alter downstream signaling events. Joubert syndrome is characterized by a specific midbrain-hindbrain malformation ('molar tooth sign'), variably associated retinal dystrophy, nephronophthisis, liver fibrosis and polydactyly and is included in the newly emerging group of 'ciliopathies'. In individuals with Joubert disease genetically linked to JBTS1, we identified mutations in the INPP5E gene, encoding inositol polyphosphate-5-phosphatase E, which hydrolyzes the 5-phosphate of PtdIns(3,4,5)P3 and PtdIns(4,5)P2. Mutations clustered in the phosphatase domain and impaired 5-phosphatase activity, resulting in altered cellular PtdIns ratios. INPP5E localized to cilia in major organs affected by Joubert syndrome, and mutations promoted premature destabilization of cilia in response to stimulation. These data link PtdIns signaling to the primary cilium, a cellular structure that is becoming increasingly recognized for its role in mediating cell signals and neuronal function.
Figures
References
-
- Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet. 2008;51:1–23. - PubMed
-
- Valente EM, et al. Distinguishing the four genetic causes of Joubert syndrome-related disorders. Ann Neurol. 2005;57:513–9. - PubMed
-
- Tsujishita Y, Guo S, Stolz LE, York JD, Hurley JH. Specificity determinants in phosphoinositide dephosphorylation: crystal structure of an archetypal inositol polyphosphate 5-phosphatase. Cell. 2001;105:379–89. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
