

ATP-sensitive K+ channel knockout induces cardiac proteome remodeling predictive of heart disease susceptibility
- PMID: 19673485
- PMCID: PMC2818626
- DOI: 10.1021/pr900561g
ATP-sensitive K+ channel knockout induces cardiac proteome remodeling predictive of heart disease susceptibility
Abstract
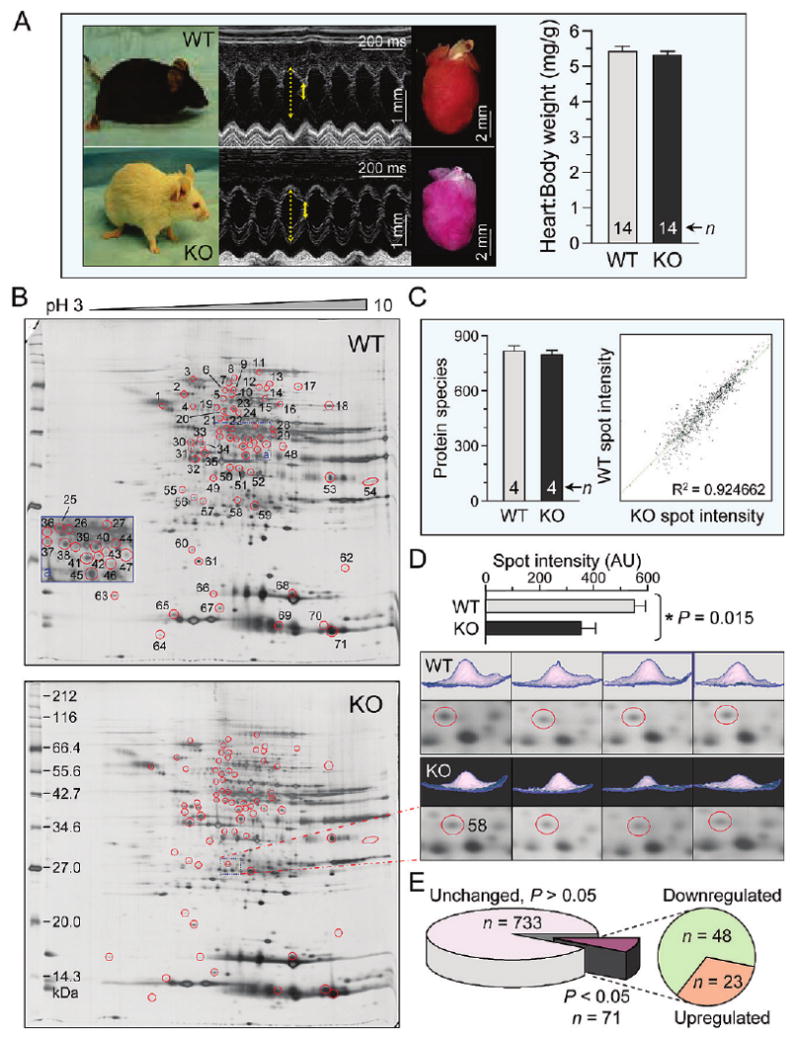
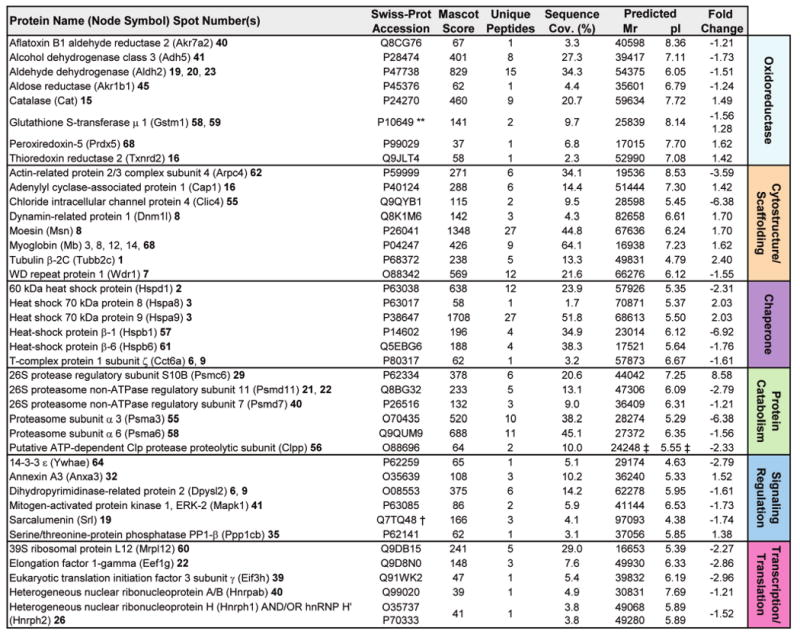
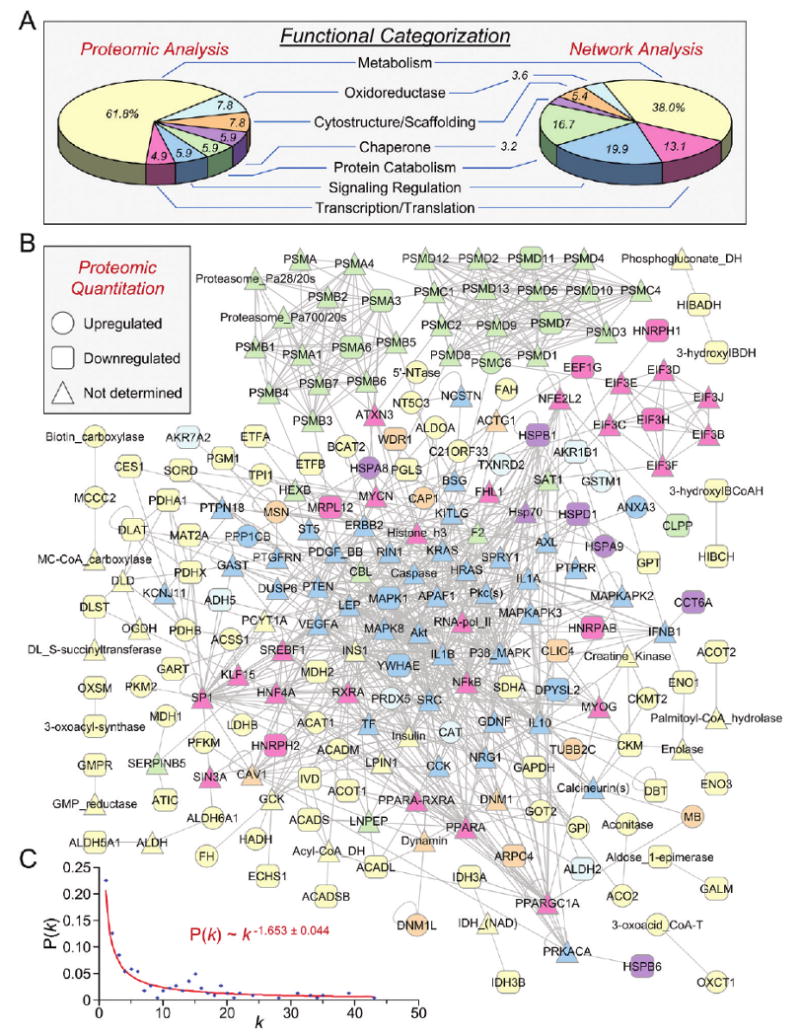
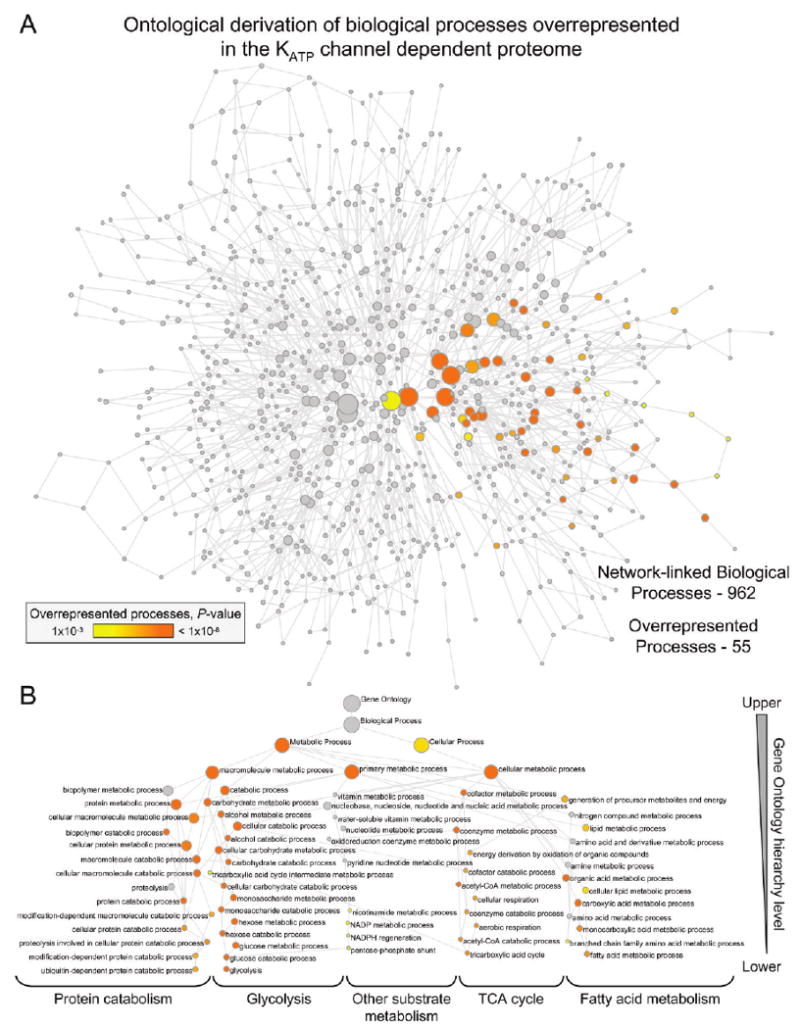
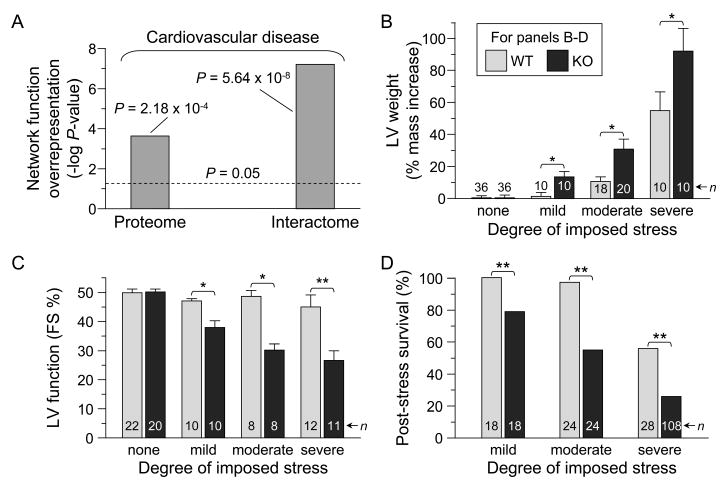
Forecasting disease susceptibility requires detection of maladaptive signatures prior to onset of overt symptoms. A case-in-point are cardiac ATP-sensitive K+ (K(ATP)) channelopathies, for which the substrate underlying disease vulnerability remains to be identified. Resolving molecular pathobiology, even for single genetic defects, mandates a systems platform to reliably diagnose disease predisposition. High-throughput proteomic analysis was here integrated with network biology to decode consequences of Kir6.2 K(ATP) channel pore deletion. Differential two-dimensional gel electrophoresis reproducibly resolved >800 protein species from hearts of asymptomatic wild-type and Kir6.2-knockout counterparts. K(ATP) channel ablation remodeled the cardiac proteome, significantly altering 71 protein spots, from which 102 unique identities were assigned following hybrid linear ion trap quadrupole-Orbitrap tandem mass spectrometry. Ontological annotation stratified the K(ATP) channel-dependent protein cohort into a predominant bioenergetic module (63 resolved identities), with additional focused sets representing signaling molecules (6), oxidoreductases (8), chaperones (6), and proteins involved in catabolism (6), cytostructure (8), and transcription and translation (5). Protein interaction mapping, in conjunction with expression level changes, localized a K(ATP) channel-associated subproteome within a nonstochastic scale-free network. Global assessment of the K(ATP) channel deficient environment verified the primary impact on metabolic pathways and revealed overrepresentation of markers associated with cardiovascular disease. Experimental imposition of graded stress precipitated exaggerated structural and functional myocardial defects in the Kir6.2-knockout, decreasing survivorship and validating the forecast of disease susceptibility. Proteomic cartography thus provides an integral view of molecular remodeling in the heart induced by K(ATP) channel deletion, establishing a systems approach that predicts outcome at a presymptomatic stage.
Figures

References
-
- Hood L, Heath JR, Phelps ME, Lin B. Systems biology and new technologies enable predictive and preventative medicine. Science. 2004;306:640–3. - PubMed
-
- Weston AD, Hood L. Systems biology, proteomics, and the future of health care: toward predictive, preventative, and personalized medicine. J Proteome Res. 2004;3:179–96. - PubMed
-
- Bell J. Predicting disease using genomics. Nature. 2004;429:453–6. - PubMed
-
- Waldman SA, Terzic A. Therapeutic targeting: a crucible for individualized medicine. Clin Pharmacol Ther. 2008;83:651–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
