

Development of epileptiform excitability in the deep entorhinal cortex after status epilepticus
- PMID: 19674083
- PMCID: PMC2776653
- DOI: 10.1111/j.1460-9568.2009.06863.x
Development of epileptiform excitability in the deep entorhinal cortex after status epilepticus
Abstract
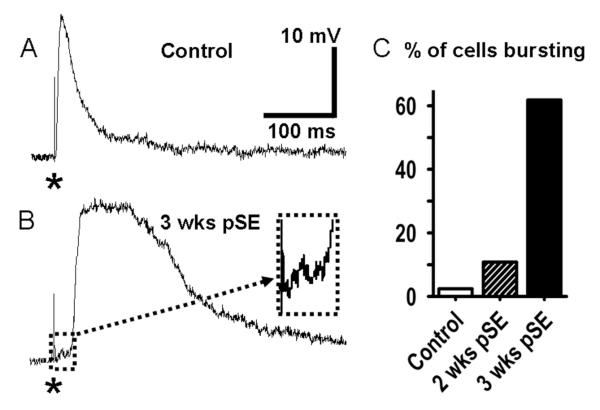
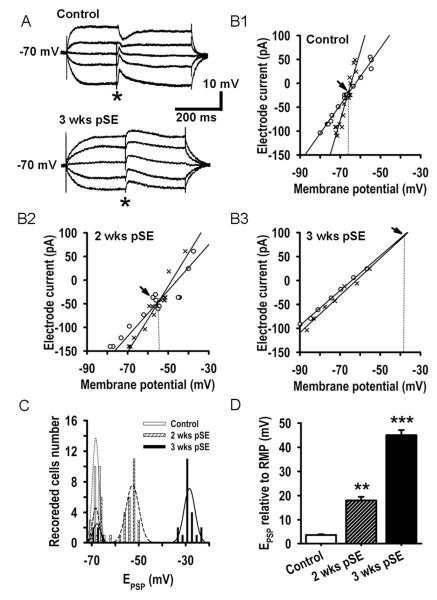
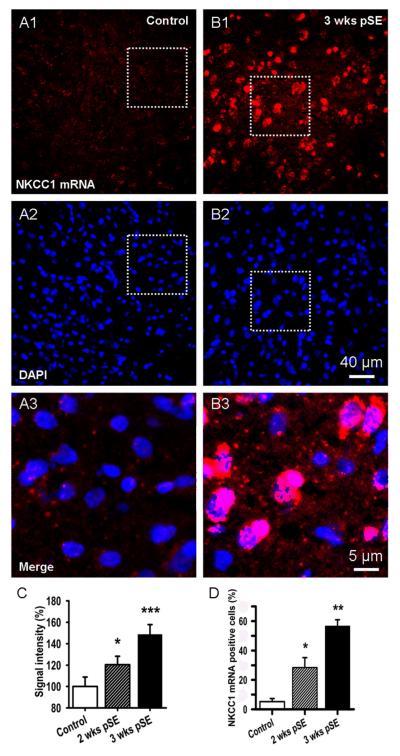
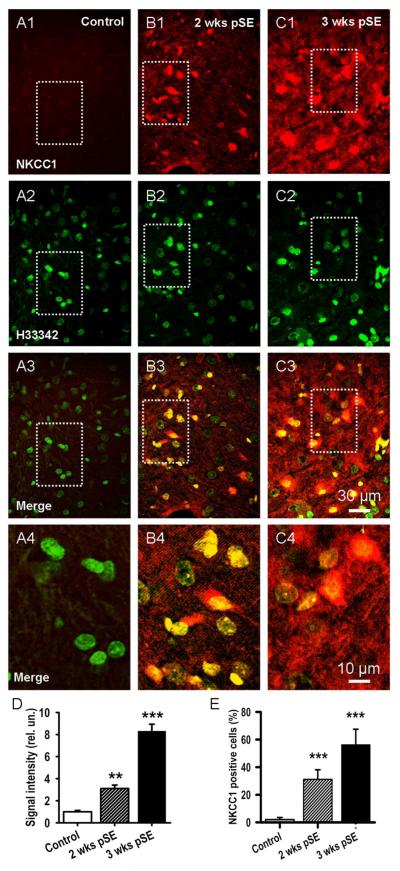
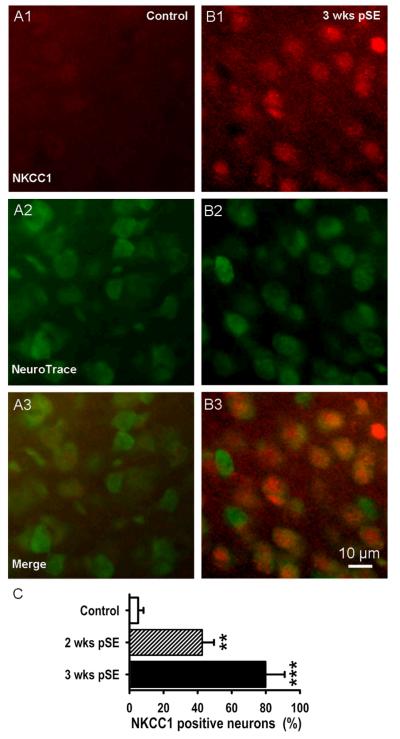
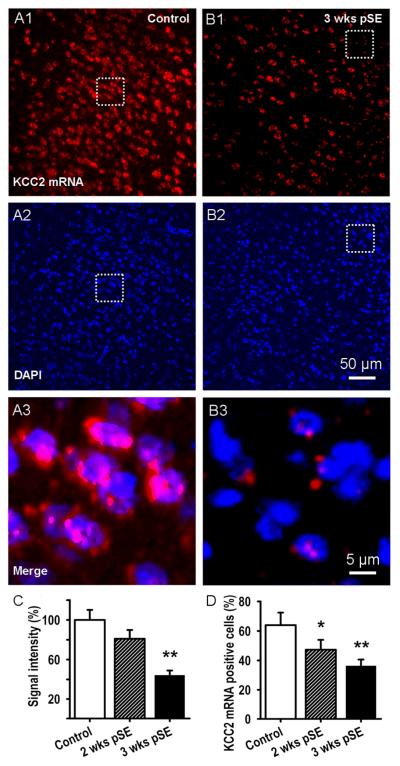
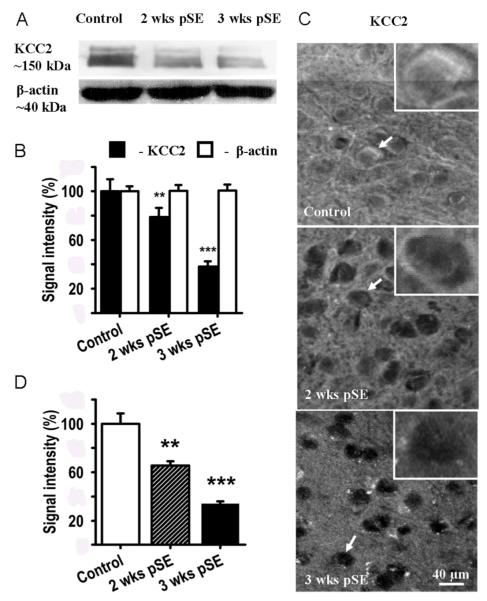
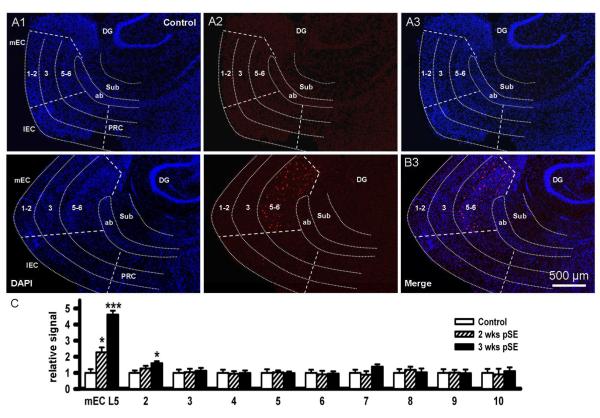
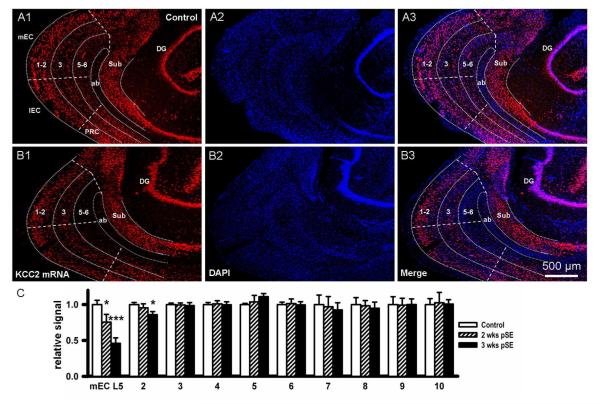
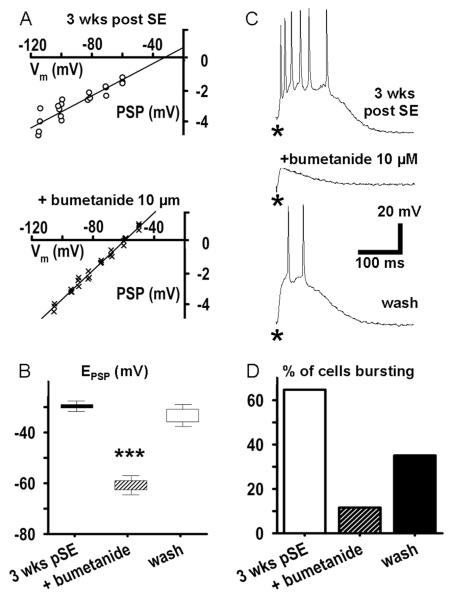
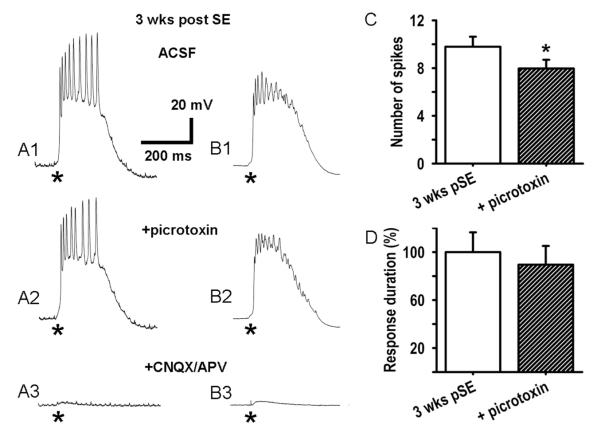
Epileptiform neuronal activity during seizures is observed in many brain areas, but its origins following status epilepticus (SE) are unclear. We have used the Li low-dose pilocarpine rat model of temporal lobe epilepsy to examine early development of epileptiform activity in the deep entorhinal cortex (EC). We show that during the 3-week latent period that follows SE, an increasing percentage of neurons in EC layer 5 respond to a single synaptic stimulus with polysynaptic burst depolarizations. This change is paralleled by a progressive depolarizing shift of the inhibitory postsynaptic potential reversal potential in layer 5 neurons, apparently caused by upregulation of the Cl(-) inward transporter NKCC1 and concurrent downregulation of the Cl(-) outward transporter KCC2, both changes favoring intracellular Cl(-) accumulation. Inhibiting Cl(-) uptake in the latent period restored more negative GABAergic reversal potentials and eliminated polysynaptic bursts. The changes in the Cl(-) transporters were highly specific to the deep EC. They did not occur in layers 1-3, perirhinal cortex, subiculum or dentate gyrus during this period. We propose that the changes in Cl(-) homeostasis facilitate hyperexcitability in the deep entorhinal cortex leading to epileptiform discharge there, which subsequently affects downstream cortical regions.
Figures
References
-
- Andre V, Dube C, Francois J, Leroy C, Rigoulot MA, Roch C, Namer IJ, Nehlig A. Pathogenesis and pharmacology of epilepsy in the lithium-pilocarpine model. Epilepsia. 2007;48(Suppl 5):41–47. - PubMed
-
- Annegers JF, Grabow JD, Groover RV, Laws ER, Jr., Elveback LR, Kurland LT. Seizures after head trauma: a population study. Neurology. 1980;30:683–689. - PubMed
-
- Bartolomei F, Khalil M, Wendling F, Sontheimer A, Regis J, Ranjeva JP, Guye M, Chauvel P. Entorhinal cortex involvement in human mesial temporal lobe epilepsy: an electrophysiologic and volumetric study. Epilepsia. 2005;46:677–687. - PubMed
-
- Bartolomei F, Wendling F, Regis J, Gavaret M, Guye M, Chauvel P. Pre-ictal synchronicity in limbic networks of mesial temporal lobe epilepsy. Epilepsy Res. 2004;61:89–104. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
