

Evaluation of clinical trials by Ethics Committees in Germany: experience of applicants with the review of requests for opinion of the Ethics Committees - results of a survey among members of the German Association of Research-Based Pharmaceutical Companies (VFA)
- PMID: 19675747
- PMCID: PMC2716553
- DOI: 10.3205/000066
Evaluation of clinical trials by Ethics Committees in Germany: experience of applicants with the review of requests for opinion of the Ethics Committees - results of a survey among members of the German Association of Research-Based Pharmaceutical Companies (VFA)
Abstract
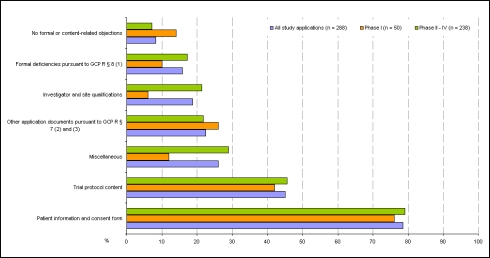
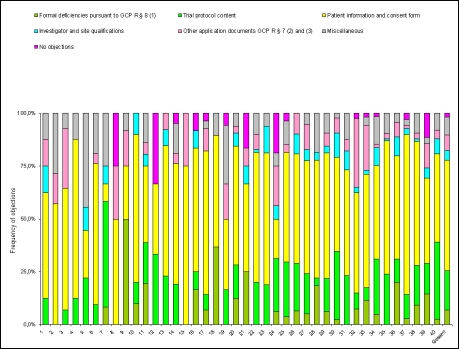
The review of requests for a positive opinion of the ethics committees (application procedure) as a requirement to start a clinical trial in Germany has been completely redesigned with the transposition of EU Directive 2001/20/EC in the 12(th) Amendment of the German Medicines Act in August 2004. The experience of applicants (sponsors, legal representatives of sponsors in the EU and persons or organizations authorized by the sponsors to make the application, respectively) in terms of interactions with the ethics committees in Germany has been positive overall, especially with respect to ethics committee adherence to the statutory timelines applicable for review of requests. However, inconsistencies between ethics committees exist in terms of the form and content of the requirements for application documents and their evaluation. With the objective of further improving both the quality of applications and the evaluation of those applications by ethics committees, a survey among members of the German Association of Research-Based Pharmaceutical Companies (VFA) was conducted from January to April 2008. Based on reasoned opinions issued by the respective ethics committee in charge of the coordinating principal investigator (coordinating ethics committee), the type and frequency of formal and content-related objections to applications according to section sign 7 of the German Good Clinical Practice (GCP) Regulation were systematically documented, and qualitative and quantitative analyses performed. 21 out of 44 members of the VFA participated in the survey. 288 applications for Phase I-IV studies submitted between January and December 2007 to 40 ethics committees were evaluated. This survey shows that about one in six applications is incomplete and has formal and/or content objections, respectively, especially those that pertain to documents demonstrating the qualification of the investigator and/or suitability of the facilities. These objections are attributable to some extent to the differing and/or unclear requirements of the individual ethics committees on the content and comprehension of the submission documents. However, applicants also need to pay more attention to the completeness and validity of the submission documents. The majority of content-related objections apply to the patient information and consent documents and study protocols submitted. Applicants on average acted upon only 3 out of 4 objections, for various reasons: the relevant information was already given in the submitted documents, but had not been taken into consideration by the ethics committees; objections were not applicable; objections lacked a legal basis. In such cases the applicants made reference to the specific information already submitted or gave reasons for not acting on the objection. This course of action was accepted by the ethics committees, with few exceptions. The survey sheds light on the existing inconsistencies in the evaluations of applications by the various ethics committees and suggests ways in which the existing constructive dialogue between applicants and ethics committees may provide a basis to further harmonize both the requirements regarding form and content of application documents, and the criteria for evaluation of applications by ethics committees within the legal framework.
Das Verfahren bei der Ethik-Kommission (EK) als Voraussetzung für den Beginn einer klinischen Prüfung, wurde in Deutschland im August 2004 mit der Umsetzung der EU-Richtlinie 2001/20/EG in der 12. AMG-Novelle grundlegend neu geregelt. Die bisherigen Erfahrungen von Antragstellern (Sponsoren, gesetzliche Vertreter von Sponsoren in der EG bzw. Personen oder Organisationen, die von den Sponsoren zur Antragstellung bevollmächtigt sind) in der Zusammenarbeit mit den Ethik-Kommissionen sind im Allgemeinen positiv, besonders was die Einhaltung der Fristen bei der Antragsbearbeitung durch die Ethik-Kommissionen betrifft. Inkonsistenzen existieren allerdings in den formalen und inhaltlichen Anforderungen an die eingereichten Antragsunterlagen und deren Bewertung durch die verschiedenen Ethik-Kommissionen.
Mit dem Ziel einer weiteren Qualitätssteigerung der Antragstellung sowie der Bewertung durch die Ethik-Kommissionen, wurden mittels einer von Januar bis April 2008 durchgeführten Umfrage bei Mitgliedsunternehmen des Verbandes Forschender Arzneimittelhersteller e.V. (VFA) systematisch Art und Häufigkeit von formalen und inhaltlichen Einwänden erfasst sowie qualitativ und quantitativ ausgewertet. Grundlage waren Bescheide der für den Leiter der klinischen Prüfung jeweils zuständigen Ethik-Kommission (federführende Ethik-Kommission) bei Erstantragstellung einer klinischen Prüfung gemäß § 7 GCP-V.
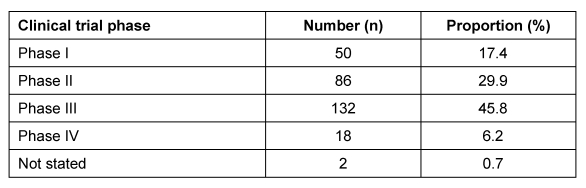
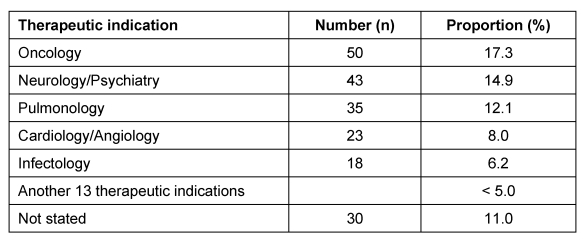
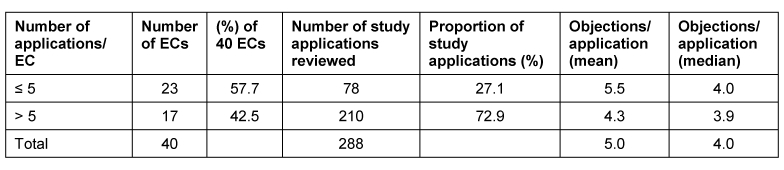
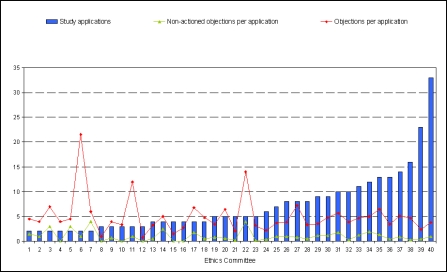
21 der 44 Mitgliedsunternehmen des VFA haben sich an der Umfrage beteiligt. Ausgewertet wurden 288 Antragsverfahren auf zustimmende Bewertung einer klinischen Prüfung der Phasen I bis IV, die im Zeitraum Januar bis Dezember 2007 bei 40 Ethik-Kommissionen gestellt wurden.
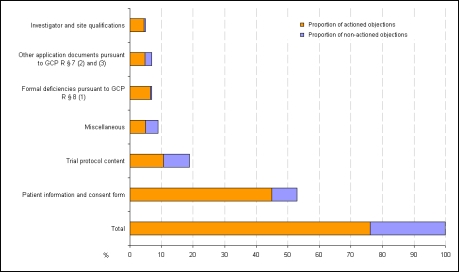
In der vorliegenden Umfrage ist etwa jeder sechste Antrag unvollständig bzw. weist formale und/oder inhaltliche Einwände zu den Antragsunterlagen, insbesondere bezüglich der Dokumentation der Prüferqualifikation und/oder Eignung der Prüfstelle auf. Diese Einwände sind zum Teil auf die unterschiedlichen und/oder unklaren Anforderungen der einzelnen Ethik-Kommissionen hinsichtlich Inhalt und Umfang der Antragsunterlagen zurückzuführen. Aber auch von Seiten der Antragsteller ist verstärkt auf die Vollständigkeit und erforderliche Aussagekraft der Antragsunterlagen zu achten. Die meisten inhaltlichen Einwände bezogen sich auf die vorgelegten Patienteninformationen und Einwilligungserklärungen sowie die Prüfpläne. Insgesamt betrachtet, sind die Antragsteller allerdings im Durchschnitt nur 3 von 4 Einwänden nachgekommen, da die von den Ethik-Kommissionen angeforderten Informationen mit den eingereichten Unterlagen bereits vorlagen, aber bei der Bewertung durch die Ethik-Kommissionen nicht berücksichtigt wurden, eine Vielzahl der Einwände nicht gerechtfertigt war oder die gesetzlichen Voraussetzungen für die Einwände fehlten. In diesen Fällen verwiesen die Antragsteller auf die vorgelegten Unterlagen oder begründeten durch nähere Erläuterungen und Stellungnahmen die Nicht-Umsetzung der Einwände. Dies wurde bis auf wenige Ausnahmen von den Ethik-Kommissionen akzeptiert. Die Umfrage beleuchtet die bestehende Heterogenität in den Bewertungen durch die verschiedenen Ethik-Kommissionen und zeigt Möglichkeiten auf, wie der bestehende konstruktive Dialog zwischen Antragsteller und Ethik-Kommissionen dazu dienen kann, die formalen und inhaltlichen Anforderungen an die einzureichenden Unterlagen sowie die Kriterien im Bewertungsverfahren klinischer Prüfungen bei den Ethik-Kommissionen innerhalb der gesetzlichen Rahmenbedingungen weiter zu harmonisieren.
Keywords: application procedure; clinical trials; ethics committees; formal and content-related objections.
Figures
References
-
- Gesetz über den Verkehr mit Arzneimitteln (Arzneimittel - AMG). Arzneimittelgesetz in der Fassung der Bekanntmachung vom 12. Dezember 2005 (BGBl. I S. 3394), das zuletzt durch Artikel 9 Abs. 1 des Gesetzes vom 23. November 2007 (BGBl. I S. 2631) geändert worden ist [German Drug Law/German Medicines Act] Berlin: Bundesministerium der Justiz; [cited 2009 Jul 07]. Available from: http://www.gesetze-im-internet.de/amg_1976/index.html.
-
- Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Official Journal of the European Union. 2001;L 121, 1/5/2001:34–44. Available from: http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-1/dir_2001_2.... - PubMed
-
- European Commission. Commission Directive 2005/28/EC of 8 April 2005 laying down principles and detailed guidelines for good clinical practice as regards investigational medicinal products for human use, as well as the requirements for authorisation of the manufacturing or importation of such products. Official Journal of the European Union. 2005;L 91, 9/4/2005:13–19. Available from: http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-1/dir_2005_2....
-
- Verordnung über die Anwendung der Guten Klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Arzneimitteln zur Anwendung am Menschen [Regulation on the implementation of Good Clinical Practice in the conduct of clinical trials on medicinal products for human use]. GCP-Verordnung vom 9. August 2004 (BGBl. I S. 2081), die zuletzt durch Artikel 4 der Verordnung vom 3. November 2006 (BGBl. I S. 2523) geändert worden ist. Berlin: Bundesministerium der Justiz; [cited 2009 Jul 07]. Available from: http://www.gesetze-im-internet.de/gcp-v/index.html.
-
- Bundesinstitut für Arzneimittel und Medizinprodukte [Statistics of the Federal Institute for Drugs and Medical Devices] Genehmigungsverfahren Statistik. Bonn: BfArM; [updated: 2008 Feb 26; cited 2009 Jul 07]. Available from: http://www.bfarm.de/cln_012/nn_1198686/DE/Arzneimittel/1__vorDerZul/kli....
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
