

Two HIV-1 variants resistant to small molecule CCR5 inhibitors differ in how they use CCR5 for entry
- PMID: 19680536
- PMCID: PMC2718843
- DOI: 10.1371/journal.ppat.1000548
Two HIV-1 variants resistant to small molecule CCR5 inhibitors differ in how they use CCR5 for entry
Abstract
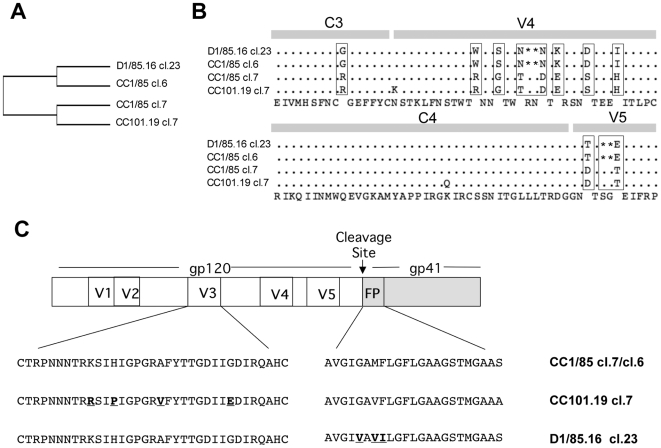
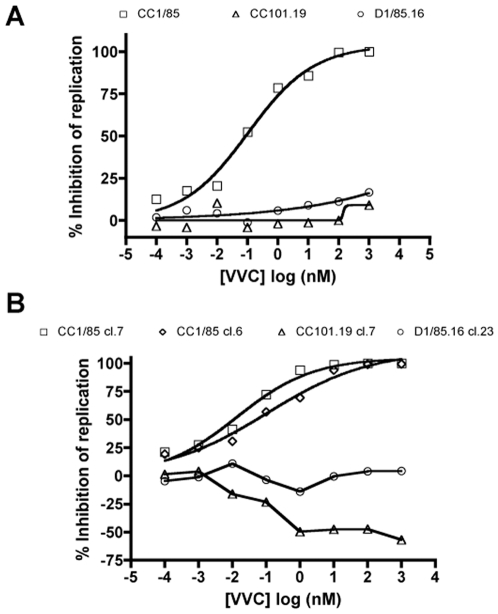
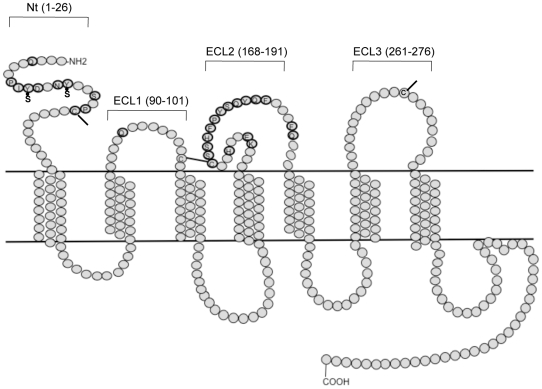
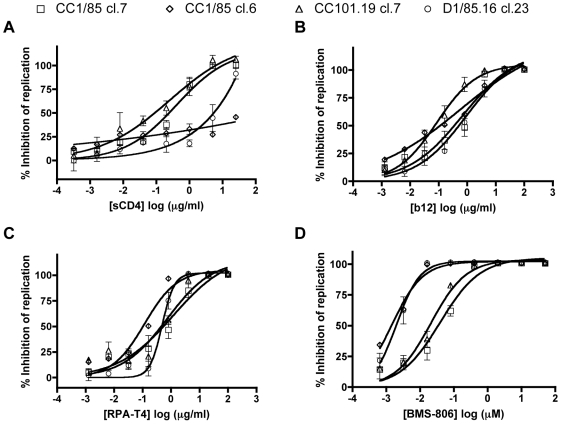
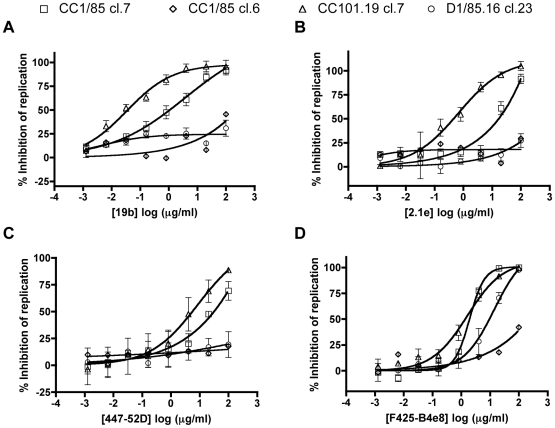
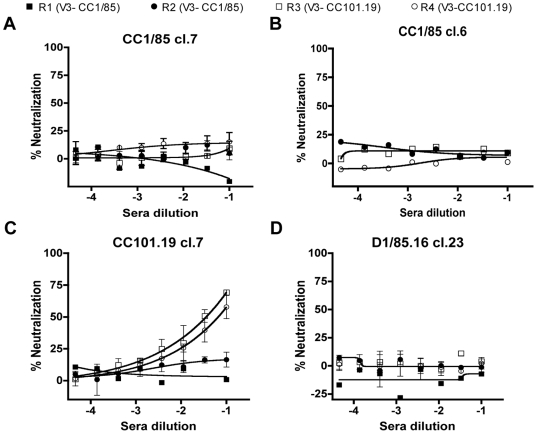
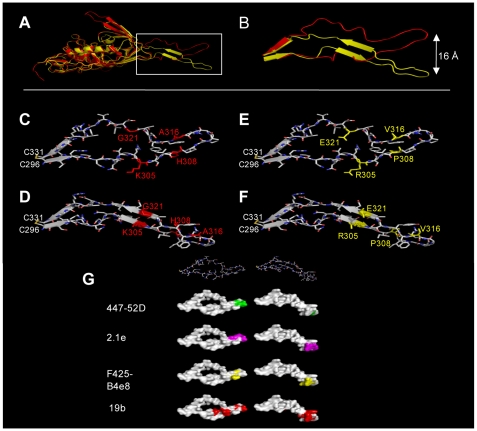
HIV-1 variants resistant to small molecule CCR5 inhibitors recognize the inhibitor-CCR5 complex, while also interacting with free CCR5. The most common genetic route to resistance involves sequence changes in the gp120 V3 region, a pathway followed when the primary isolate CC1/85 was cultured with the AD101 inhibitor in vitro, creating the CC101.19 resistant variant. However, the D1/86.16 escape mutant contains no V3 changes but has three substitutions in the gp41 fusion peptide. By using CCR5 point-mutants and gp120-targeting agents, we have investigated how infectious clonal viruses derived from the parental and both resistant isolates interact with CCR5. We conclude that the V3 sequence changes in CC101.19 cl.7 create a virus with an increased dependency on interactions with the CCR5 N-terminus. Elements of the CCR5 binding site associated with the V3 region and the CD4-induced (CD4i) epitope cluster in the gp120 bridging sheet are more exposed on the native Env complex of CC101.19 cl.7, which is sensitive to neutralization via these epitopes. However, D1/86.16 cl.23 does not have an increased dependency on the CCR5 N-terminus, and its CCR5 binding site has not become more exposed. How this virus interacts with the inhibitor-CCR5 complex remains to be understood.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
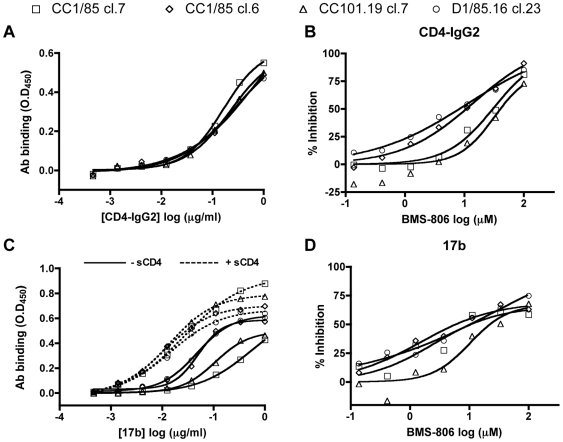
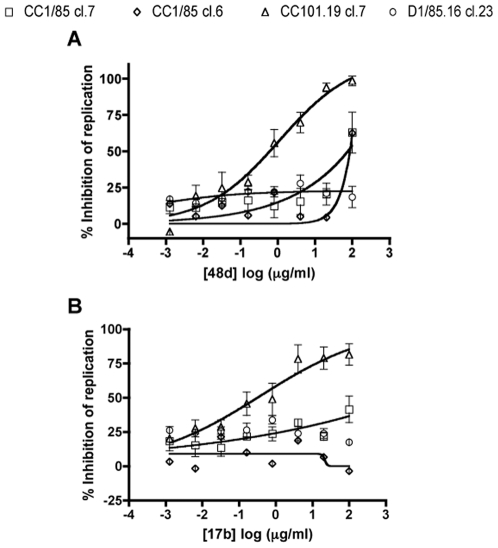
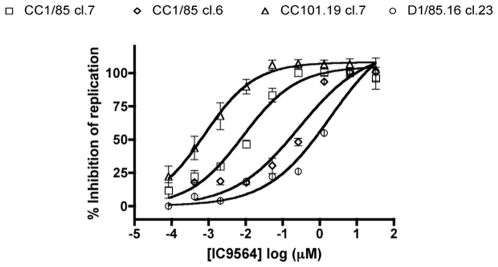
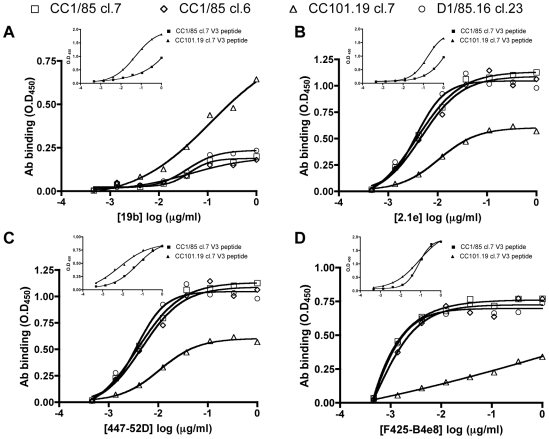
References
-
- Kuhmann SE, Hartley O. Targeting chemokine receptors in HIV: a status report. Annu Rev Pharmacol Toxicol. 2008;48:425–461. - PubMed
-
- Ray N, Doms RW. HIV-1 coreceptors and their inhibitors. Curr Top Microbiol Immunol. 2006;303:97–120. - PubMed
-
- Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, et al. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol Pharmacol. 2008;73:789–800. - PubMed
-
- Seibert C, Ying W, Gavrilov S, Tsamis F, Kuhmann SE, et al. Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology. 2006;349:41–54. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
