

The 1.9 a structure of human alpha-N-acetylgalactosaminidase: The molecular basis of Schindler and Kanzaki diseases
- PMID: 19683538
- PMCID: PMC2771859
- DOI: 10.1016/j.jmb.2009.08.021
The 1.9 a structure of human alpha-N-acetylgalactosaminidase: The molecular basis of Schindler and Kanzaki diseases
Abstract
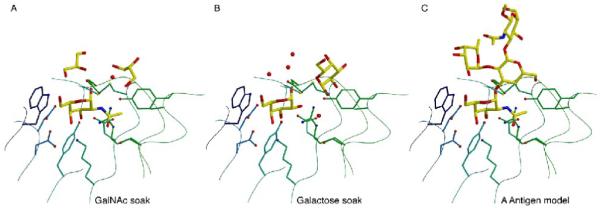
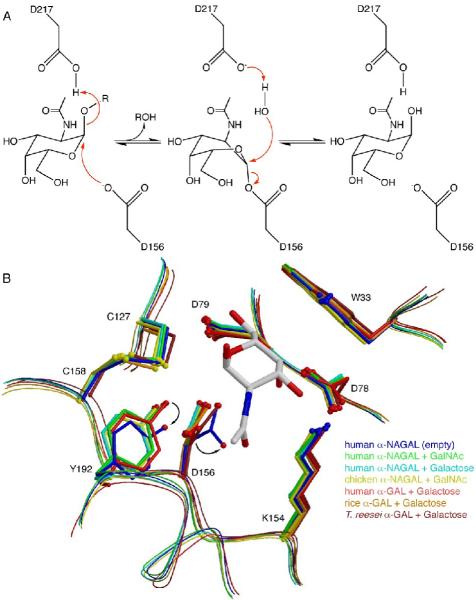
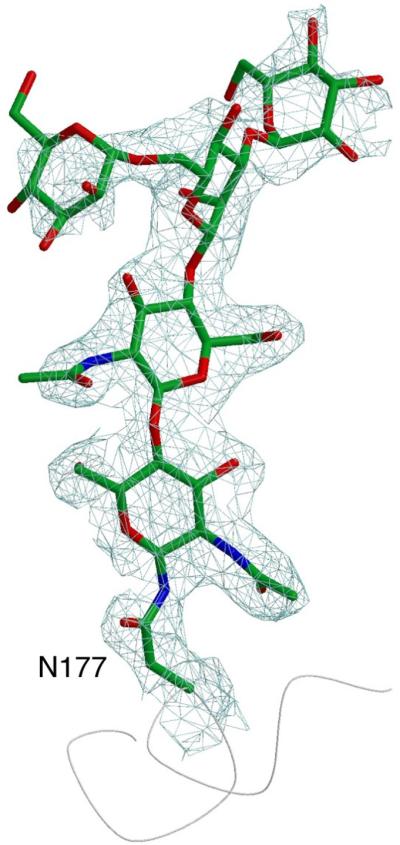
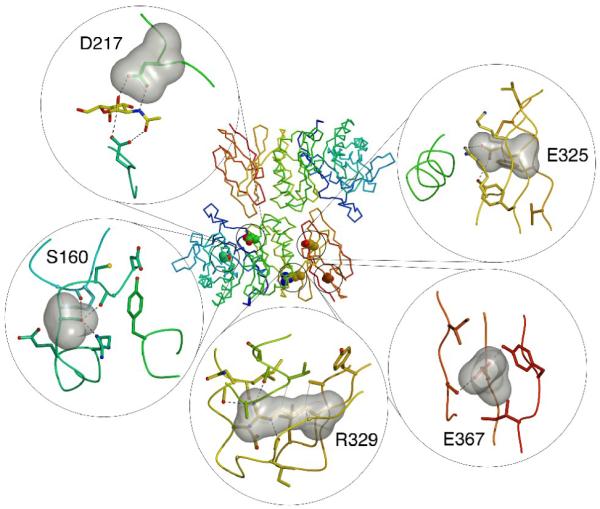
alpha-N-acetylgalactosaminidase (alpha-NAGAL; E.C. 3.2.1.49) is a lysosomal exoglycosidase that cleaves terminal alpha-N-acetylgalactosamine residues from glycopeptides and glycolipids. In humans, a deficiency of alpha-NAGAL activity results in the lysosomal storage disorders Schindler disease and Kanzaki disease. To better understand the molecular defects in the diseases, we determined the crystal structure of human alpha-NAGAL after expressing wild-type and glycosylation-deficient glycoproteins in recombinant insect cell expression systems. We measured the enzymatic parameters of our purified wild-type and mutant enzymes, establishing their enzymatic equivalence. To investigate the binding specificity and catalytic mechanism of the human alpha-NAGAL enzyme, we determined three crystallographic complexes with different catalytic products bound in the active site of the enzyme. To better understand how individual defects in the alpha-NAGAL glycoprotein lead to Schindler disease, we analyzed the effect of disease-causing mutations on the three-dimensional structure.
Figures
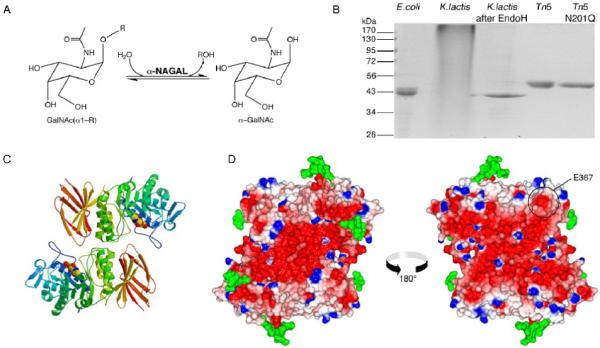
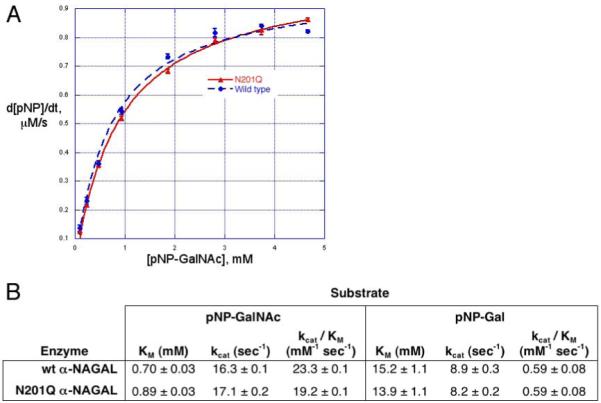
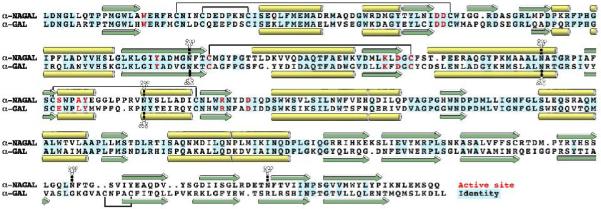
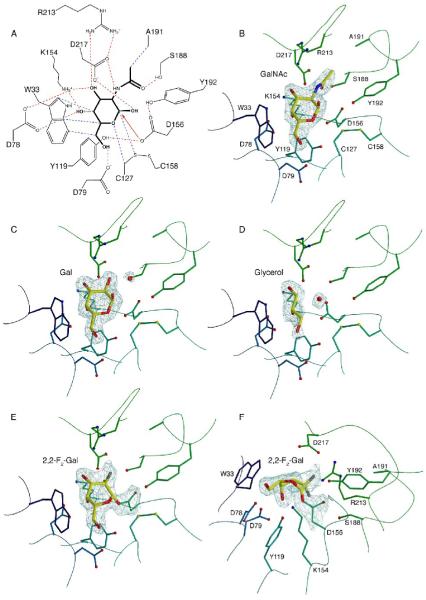
References
-
- van Diggelen OP, Schindler D, Kleijer WJ, Huijmans JM, Galjaard H, Linden HU, Peter-Katalinic J, Egge H, Dabrowski U, Cantz M. Lysosomal α-N-acetylgalactosaminidase deficiency: a new inherited metabolic disease. Lancet. 1987;2:804. - PubMed
-
- Schindler D, Bishop DF, Wolfe DE, Wang AM, Egge H, Lemieux RU, Desnick RJ. Neuroaxonal dystrophy due to lysosomal α-N-acetylgalactosaminidase deficiency. N. Engl. J. Med. 1989;320:1735–40. - PubMed
-
- Desnick RJ, Schindler D. α-N-Acetylgalactosaminidase Deficiency: Schindler Disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edit. McGraw-Hill; New York: 2001. pp. 3483–3505.
-
- van Diggelen OP, Schindler D, Willemsen R, Boer M, Kleijer WJ, Huijmans JG, Blom W, Galjaard H. α-N-acetylgalactosaminidase deficiency, a new lysosomal storage disorder. J. Inherit. Metab. Dis. 1988;11:349–57. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
