

Nuclear EGFR contributes to acquired resistance to cetuximab
- PMID: 19684613
- PMCID: PMC2900381
- DOI: 10.1038/onc.2009.234
Nuclear EGFR contributes to acquired resistance to cetuximab
Abstract
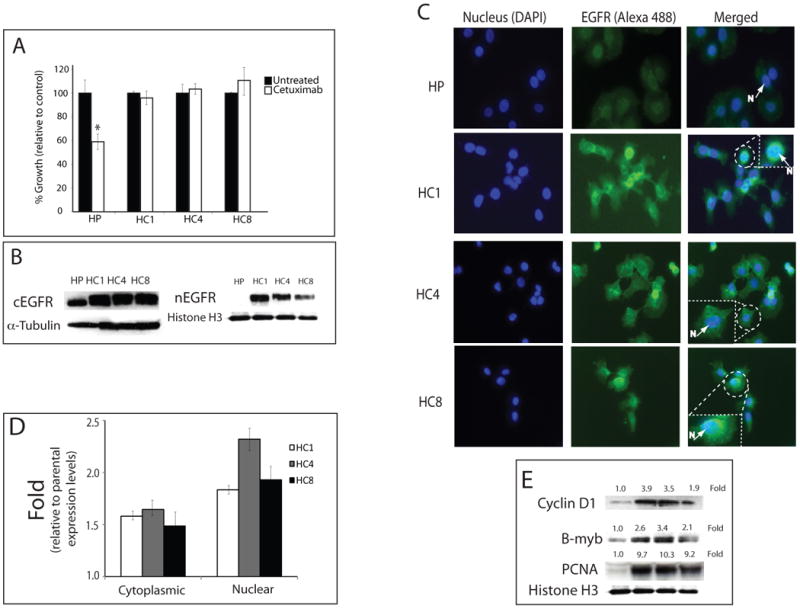
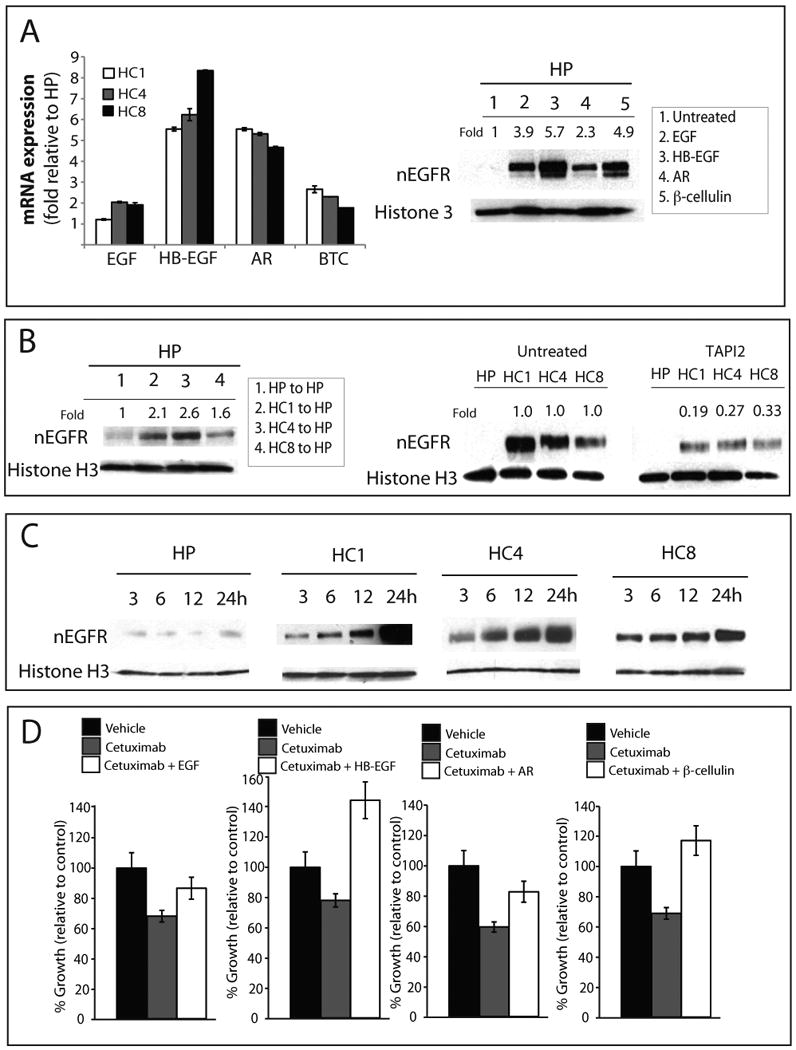
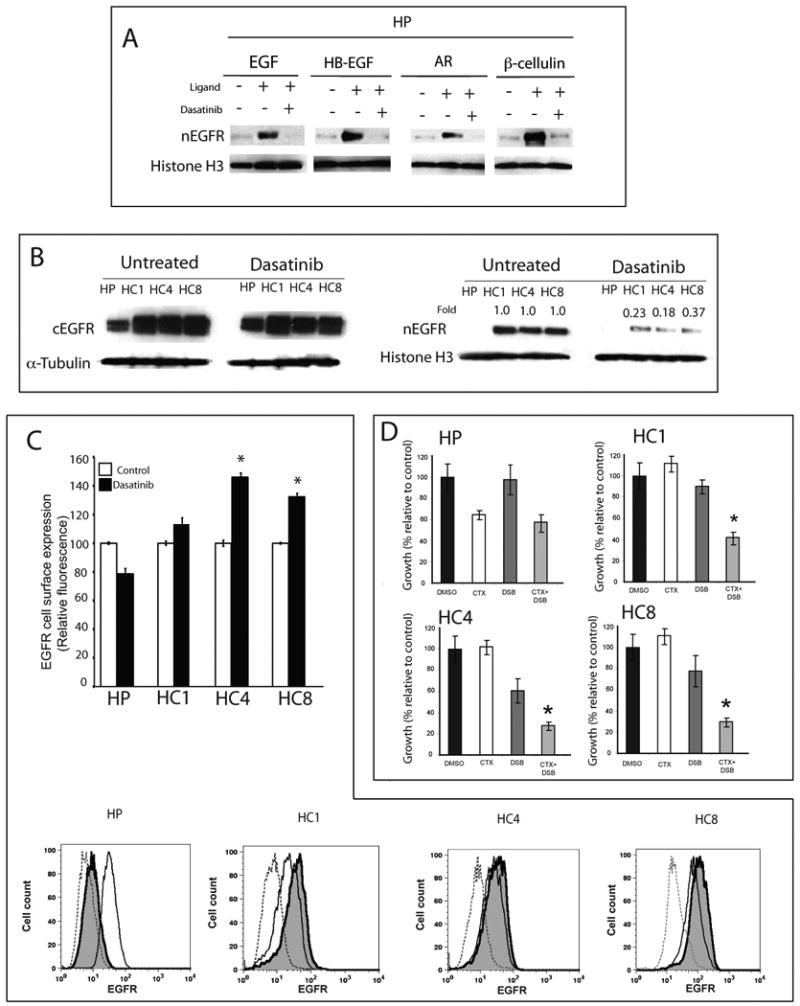
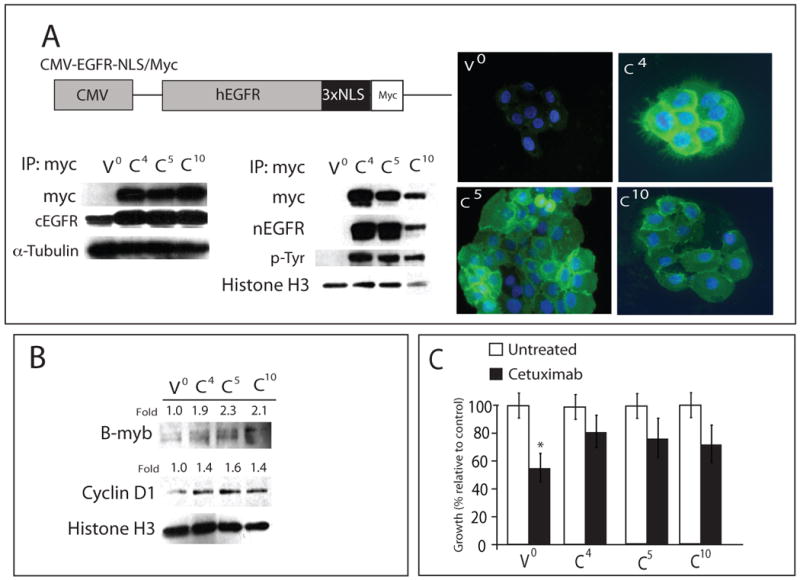
Epidermal growth factor receptor (EGFR) is a ubiquitously expressed receptor tyrosine kinase involved in the etiology of several human cancers. Cetuximab is an EGFR-blocking antibody that has been approved for the treatment of patients with head and neck squamous cell carcinoma and metastatic colorectal cancer. Previous reports have shown that EGFR translocation to the nucleus is associated with cell proliferation. Here we investigated mechanisms of acquired resistance to cetuximab using a model derived from the non-small cell lung cancer line H226. We demonstrated that cetuximab-resistant cells overexpress HER family ligands including epidermal growth factor (EGF), amphiregulin, heparin-binding EGF and beta-cellulin. Overexpression of these ligands is associated with the nuclear translocation of the EGFR and this process was mediated by the Src family kinases (SFK). Treatment of cetuximab-resistant cells with the SFK inhibitor, dasatinib, resulted in loss of nuclear EGFR, increased membrane expression of the EGFR and resensitization to cetuximab. In addition, expression of a nuclear localization sequence-tagged EGFR in cetuximab-sensitive cells increased resistance to cetuximab both in vitro and in mouse xenografts. Collectively, these data suggest that nuclear expression of EGFR may be an important molecular determinant of resistance to cetuximab therapy and provides a rationale for investigating nuclear EGFR as a biomarker for cetuximab response. Further, these data suggest a rationale for the design of clinical trials that examine the value of treating patients with cetuximab-resistant tumors with inhibitors of SFKs in combination with cetuximab.
Conflict of interest statement
Figures
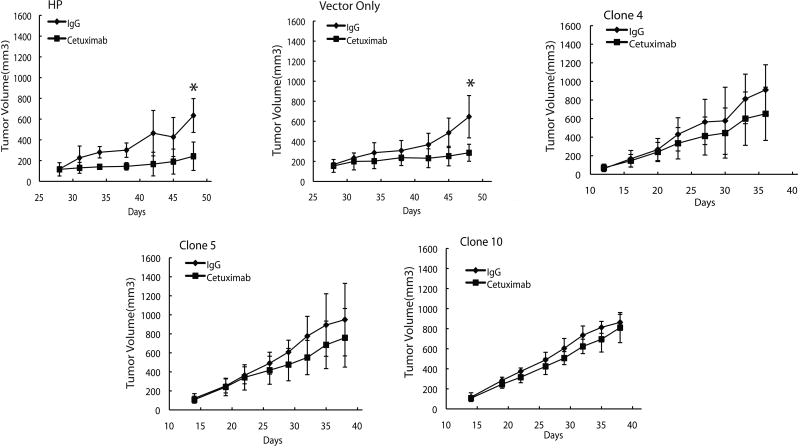
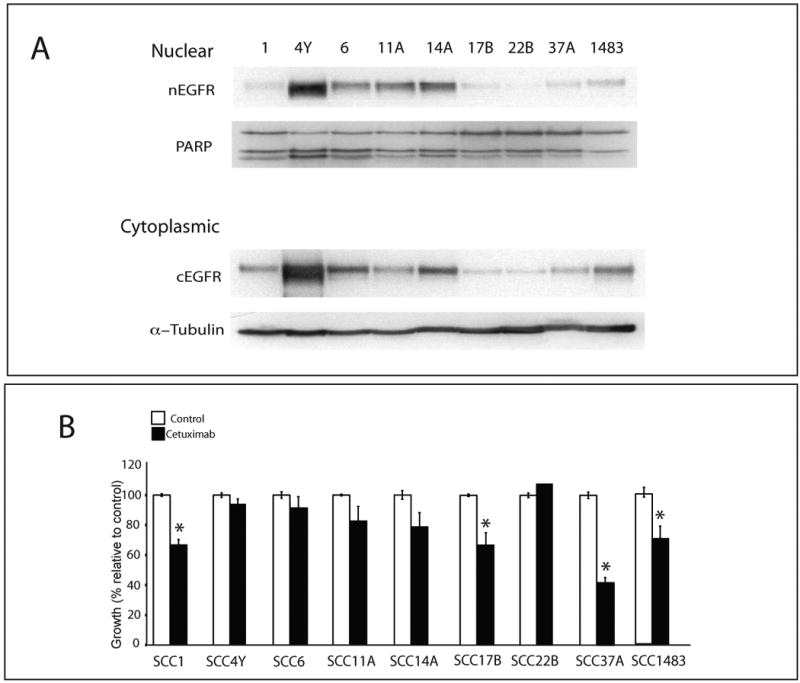
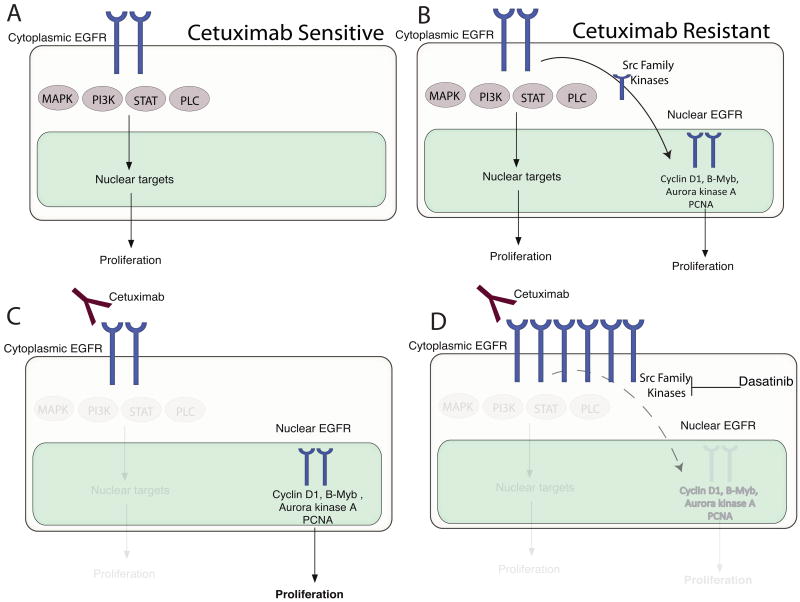
References
-
- Arteaga CL. EGF receptor as a therapeutic target: patient selection and mechanisms of resistance to receptor-targeted drugs. J Clin Oncol. 2003;21:289s–291s. - PubMed
-
- Bandyopadhyay D, Mandal M, Adam L, Mendelsohn J, Kumar R. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J Biol Chem. 1998;273:1568–73. - PubMed
-
- Bianco R, Troiani T, Tortora G, Ciardiello F. Intrinsic and acquired resistance to EGFR inhibitors in human cancer therapy. Endocr Relat Cancer. 2005;12:S159–171. - PubMed
-
- Camp ER, Summy J, Bauer TW, Liu W, Gallick GE, Ellis LM. Molecular Mechanisms of Resistance to Therapies Targeting the Epidermal Growth Factor Receptor. Clin Cancer Res. 2005;11:397–405. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
