

How the fanconi anemia pathway guards the genome
- PMID: 19686080
- PMCID: PMC2830711
- DOI: 10.1146/annurev-genet-102108-134222
How the fanconi anemia pathway guards the genome
Abstract
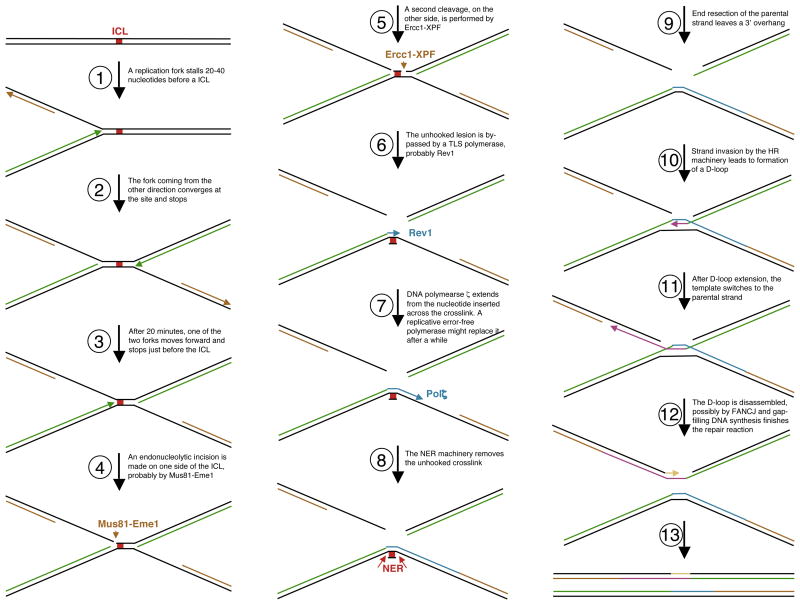
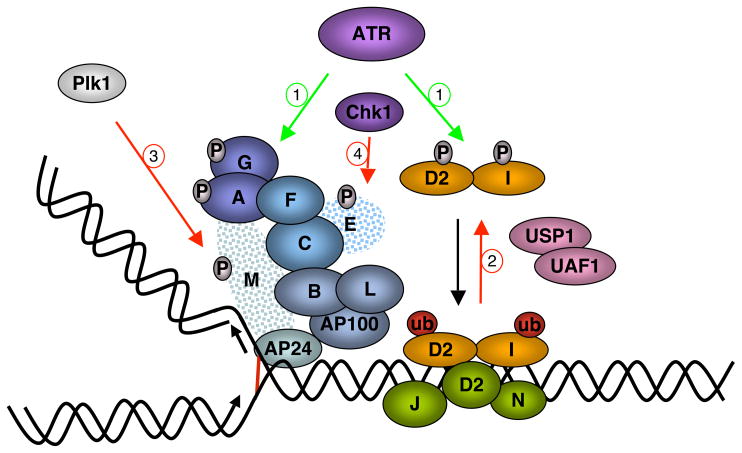
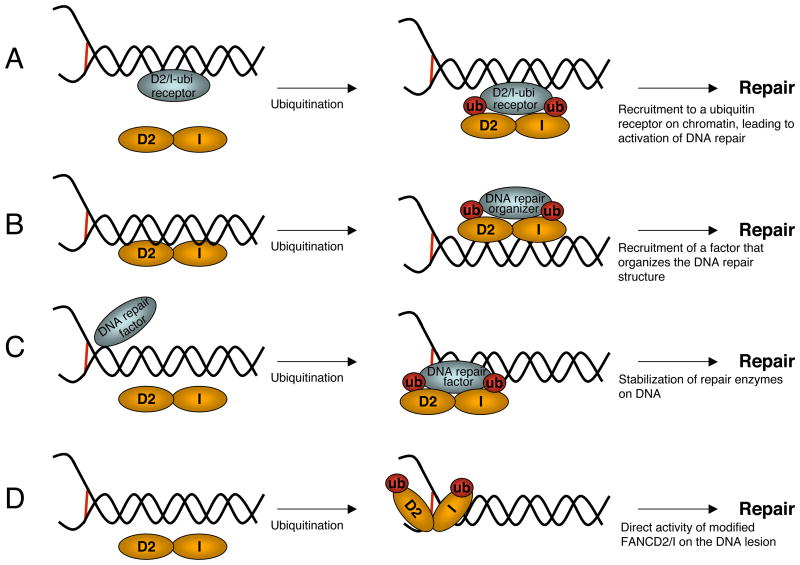
Fanconi Anemia (FA) is an inherited genomic instability disorder, caused by mutations in genes regulating replication-dependent removal of interstrand DNA crosslinks. The Fanconi Anemia pathway is thought to coordinate a complex mechanism that enlists elements of three classic DNA repair pathways, namely homologous recombination, nucleotide excision repair, and mutagenic translesion synthesis, in response to genotoxic insults. To this end, the Fanconi Anemia pathway employs a unique nuclear protein complex that ubiquitinates FANCD2 and FANCI, leading to formation of DNA repair structures. Lack of obvious enzymatic activities among most FA members has made it challenging to unravel its precise modus operandi. Here we review the current understanding of how the Fanconi Anemia pathway components participate in DNA repair and discuss the mechanisms that regulate this pathway to ensure timely, efficient, and correct restoration of chromosomal integrity.
Figures

References
-
- Agoulnik AI, Lu B, Zhu Q, Truong C, Ty MT, et al. A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum Mol Genet. 2002;11:3047–53. - PubMed
-
- Alpi AF, Pace PE, Babu MM, Patel KJ. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell. 2008;32:767–77. - PubMed
-
- Alter BP. Fanconi’s anemia and malignancies. Am J Hematol. 1996;53:99–110. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
