

Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity
- PMID: 19687229
- PMCID: PMC2737165
- DOI: 10.1084/jem.20082392
Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity
Abstract
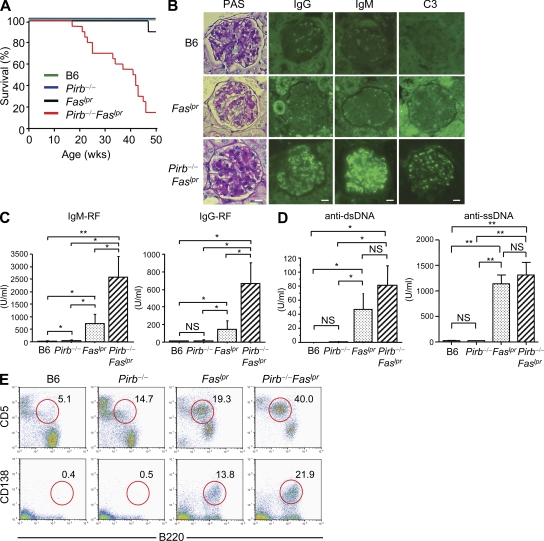
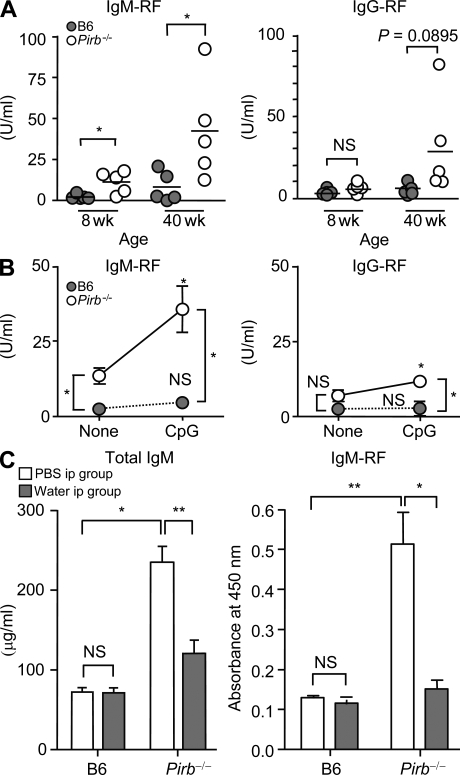
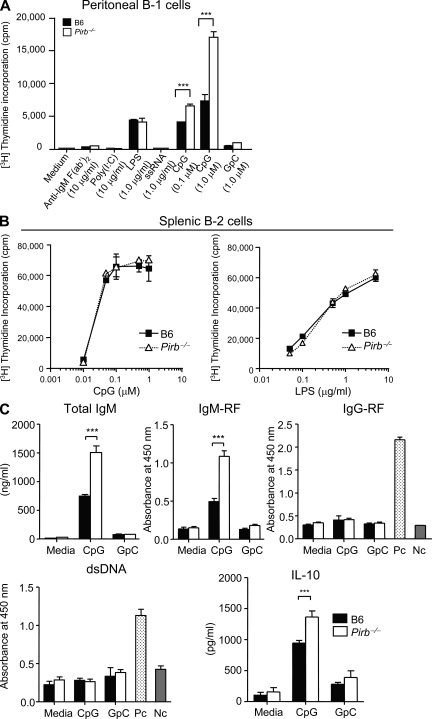
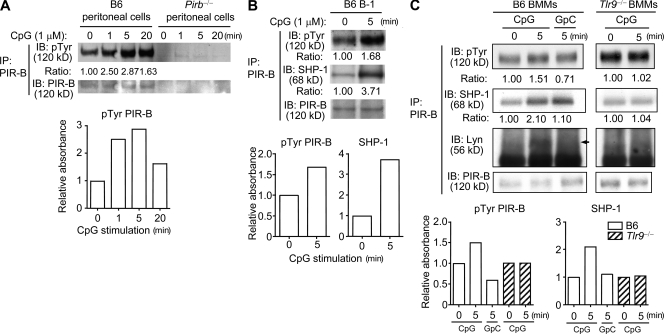
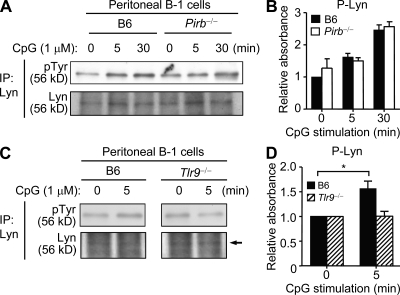
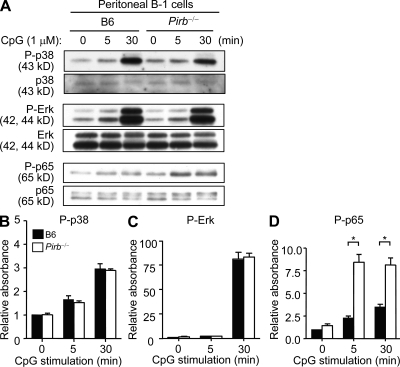
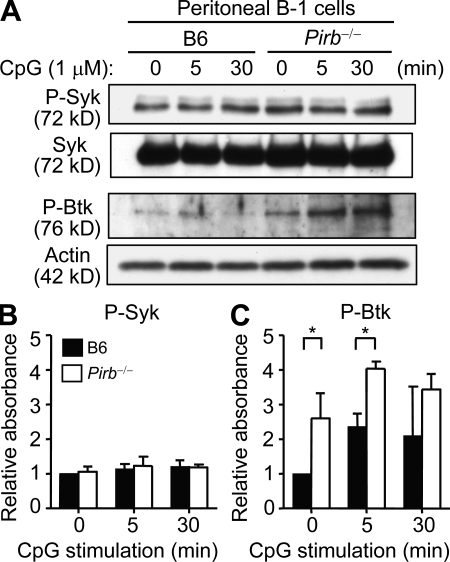
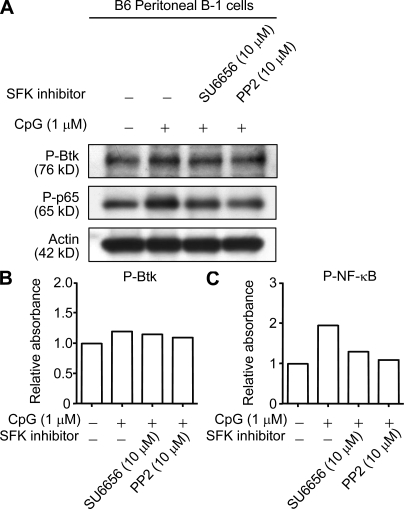
Pathogens are sensed by Toll-like receptors (TLRs) expressed in leukocytes in the innate immune system. However, excess stimulation of TLR pathways is supposed to be connected with provocation of autoimmunity. We show that paired immunoglobulin (Ig)-like receptor B (PIR-B), an immunoreceptor tyrosine-based inhibitory motif-harboring receptor for major histocompatibility class I molecules, on relatively primitive B cells, B-1 cells, suppresses TLR9 signaling via Bruton's tyrosine kinase (Btk) dephosphorylation, which leads to attenuated activation of nuclear factor kappaB p65RelA but not p38 or Erk, and blocks the production of natural IgM antibodies, including anti-IgG Fc autoantibodies, particularly rheumatoid factor. The autoantibody production in PIR-B-deficient (Pirb(-/-)) mice was further augmented in combination with the Fas(lpr) mutation, which might be linked to the development of autoimmune glomerulonephritis. These results show the critical link between TLR9-mediated sensing and a simultaneously evoked, PIR-B-mediated inhibitory circuit with a Btk intersection in B-1 cells, and suggest a novel way toward preventing pathogenic natural autoantibody production.
Figures
References
-
- Akira S., Takeda K., Kaisho T. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity.Nat. Immunol. 2:675–680 - PubMed
-
- An H., Hou J., Zhou J., Zhao W., Xu H., Zheng Y., Yu Y., Liu S., Cao X. 2008. Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1.Nat. Immunol. 9:542–550 - PubMed
-
- Baccala R., Hoebe K., Kono D.H., Beutler B., Theofilopoulos A.N. 2007. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity.Nat. Med. 13:543–551 - PubMed
-
- Baumgarth N., Tung J.W., Herzenberg L.A. 2005. B-1 B cells: development, selection, natural autoantibody and leukemia.Springer Semin. Immunopathol. 26:347–362 - PubMed
-
- Berland R., Wortis H.H. 2002. Origins and functions of B-1 cells with notes on the role of CD5.Annu. Rev. Immunol. 20:253–300 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
