

What have we learned from the congenital myasthenic syndromes
- PMID: 19688192
- PMCID: PMC3050586
- DOI: 10.1007/s12031-009-9229-0
What have we learned from the congenital myasthenic syndromes
Abstract
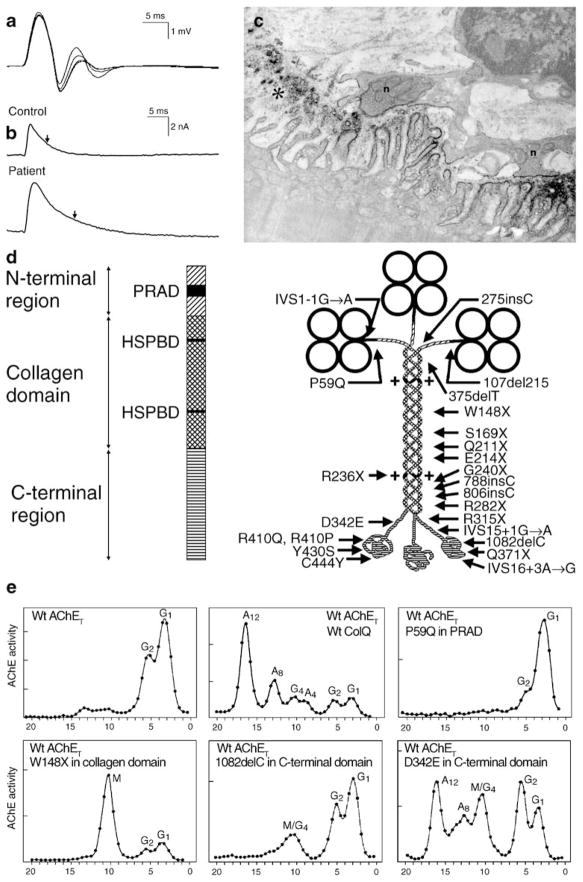
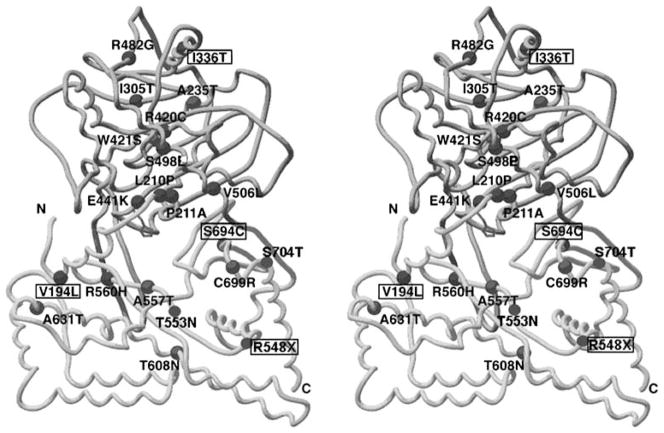
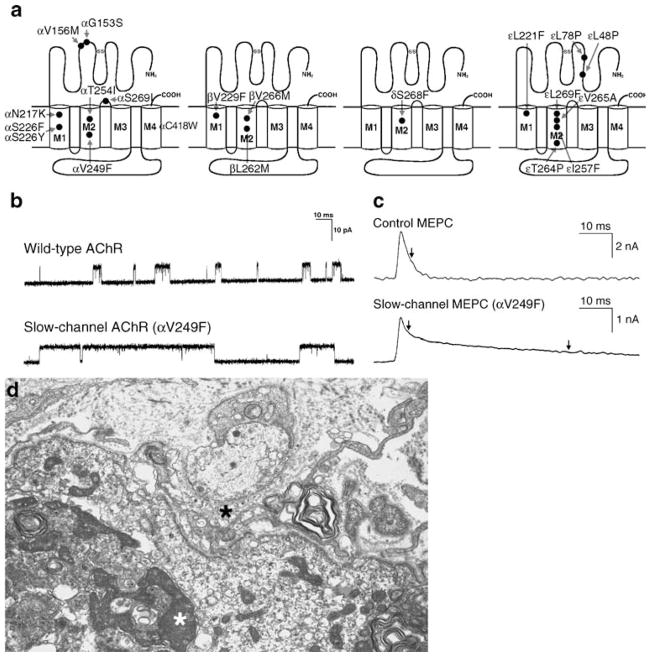
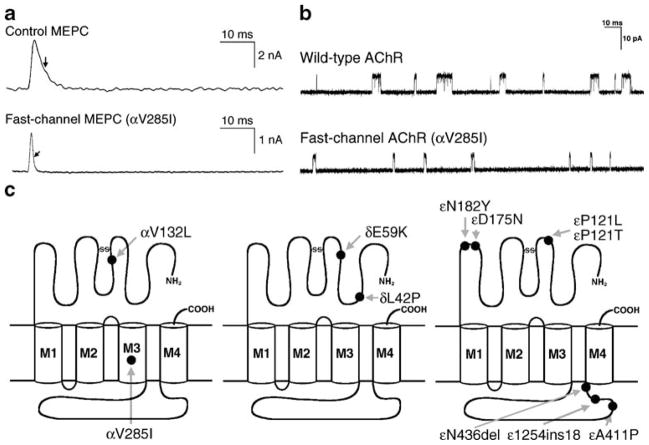
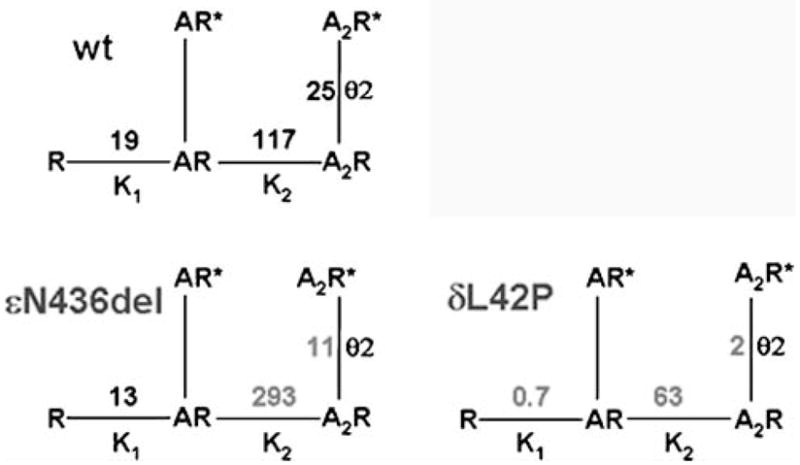
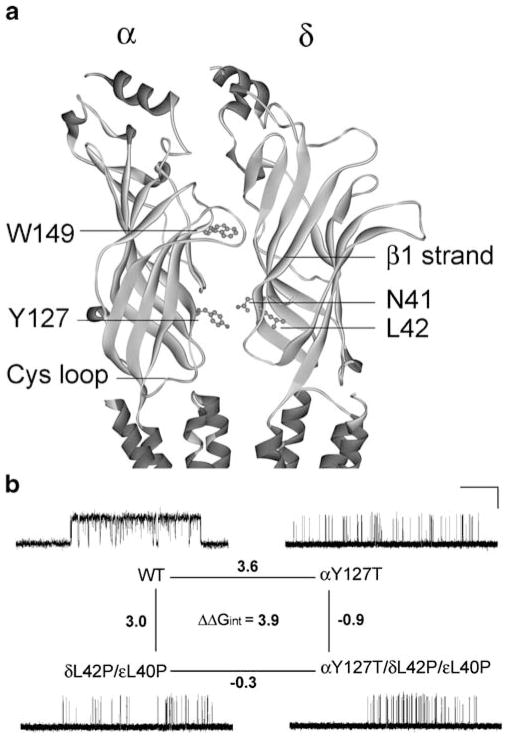
The congenital myasthenic syndromes have now been traced to an array of molecular targets at the neuromuscular junction encoded by no fewer than 11 disease genes. The disease genes were identified by the candidate gene approach, using clues derived from clinical, electrophysiological, cytochemical, and ultrastructural features. For example, electrophysiologic studies in patients suffering from sudden episodes of apnea pointed to a defect in acetylcholine resynthesis and CHAT as the candidate gene (Ohno et al., Proc Natl Acad Sci USA 98:2017-2022, 2001); refractoriness to anticholinesterase medications and partial or complete absence of acetylcholinesterase (AChE) from the endplates (EPs) has pointed to one of the two genes (COLQ and ACHE ( T )) encoding AChE, though mutations were observed only in COLQ. After a series of patients carrying mutations in a disease gene have been identified, the emerging genotype-phenotype correlations provided clues for targeted mutation analysis in other patients. Mutations in EP-specific proteins also prompted expression studies that proved pathogenicity, highlighted important functional domains of the abnormal proteins, and pointed to rational therapy.
Figures
References
-
- Barisic N, Muller JS, Paucic-Kirincic E, et al. Clinical variability of CMS-EA (congenital myasthenic syndrome with episodic apnea) due to identical CHAT mutations in two infants. European Journal of Paediatric Neurology. 2005;9:7–12. - PubMed
-
- Beeson D, Hantai D, Lochmuller H, Engel AG. Report of the 126th International Workshop: Congenital myasthenic syndromes. Neuromuscular Disorders. 2005;15:498–512. - PubMed
-
- Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. - PubMed
-
- Bon S, Coussen F, Massoulié J. Quaternary associations of acetylcholinesterase. II. The polyproline attachment domain of the collagen tail. Journal of Biological Chemistry. 1997;272:3016–3021. - PubMed
-
- Byring RF, Pihko H, Shen XM, et al. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscular Disorders. 2002;12:548–553. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
