

The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance
- PMID: 19690169
- PMCID: PMC2788836
- DOI: 10.1074/jbc.M109.000950
The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance
Abstract
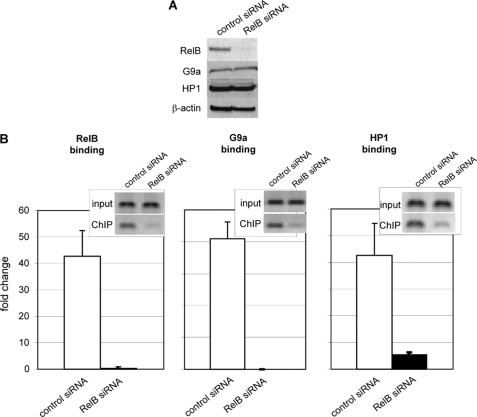
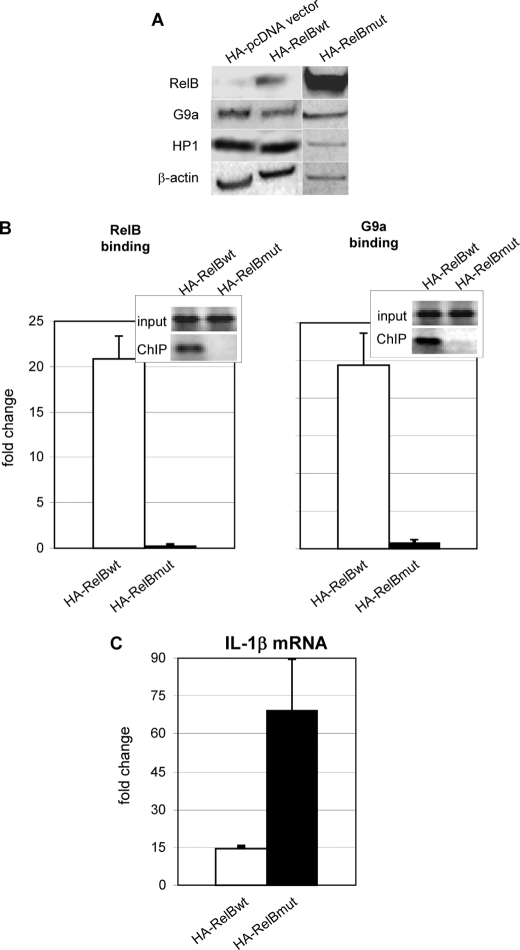
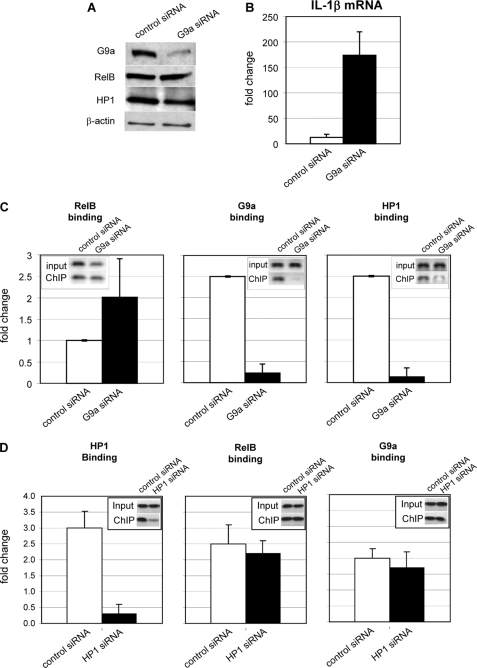
The interplay of transcription factors, histone modifiers, and DNA modification can alter chromatin structure that epigenetically controls gene transcription. During severe systemic inflammatory (SSI), the generation of facultative heterochromatin from euchromatin reversibly silences transcription of a set of acute proinflammatory genes. This gene-specific silencing is a salient feature of the endotoxin tolerant phenotype that is found in blood leukocytes of SSI patients and in a human THP-1 cell model of SSI. We previously reported that de novo induction of the NF-kappaB transcription factor RelB by endotoxin activation is necessary and sufficient for silencing transcription of acute proinflammatory genes in the endotoxin tolerant SSI phenotype. Here, we examined how RelB silences gene expression and found that RelB induces facultative heterochromatin formation by directly interacting with the histone H3 lysine 9 methyltransferase G9a. We found that heterochromatin protein 1 (HP1) and G9a formed a complex at the interleukin-1beta promoter that is dependent on the Rel homology domain (RHD) of RelB. RelB knockdown disassociated the complex and reversed transcription silencing. We also observed that whereas RelB chromatin binding was independent of G9a, RelB transcriptional silencing required G9a accumulation at the silenced promoter. Binding between RelB and G9a was confirmed by glutathione S-transferase pulldown in vitro and coimmunoprecipitation in vivo. These data provide novel insight into how RelB is required to initiate silencing in the phenotype associated with severe systemic inflammation in humans, a disease with major morbidity and mortality.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
