

Polarizable atomic multipole X-ray refinement: application to peptide crystals
- PMID: 19690373
- PMCID: PMC2733883
- DOI: 10.1107/S0907444909022707
Polarizable atomic multipole X-ray refinement: application to peptide crystals
Abstract
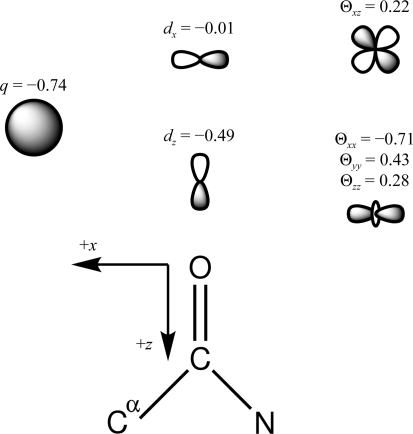
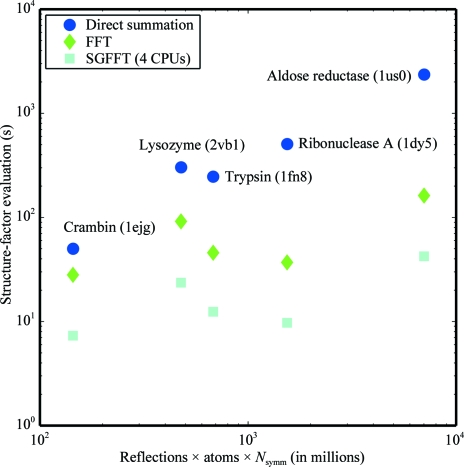
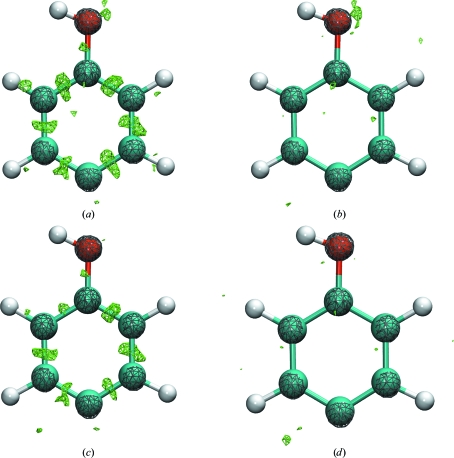
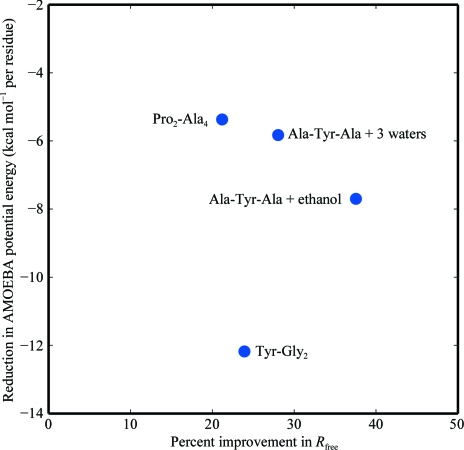
Recent advances in computational chemistry have produced force fields based on a polarizable atomic multipole description of biomolecular electrostatics. In this work, the Atomic Multipole Optimized Energetics for Biomolecular Applications (AMOEBA) force field is applied to restrained refinement of molecular models against X-ray diffraction data from peptide crystals. A new formalism is also developed to compute anisotropic and aspherical structure factors using fast Fourier transformation (FFT) of Cartesian Gaussian multipoles. Relative to direct summation, the FFT approach can give a speedup of more than an order of magnitude for aspherical refinement of ultrahigh-resolution data sets. Use of a sublattice formalism makes the method highly parallelizable. Application of the Cartesian Gaussian multipole scattering model to a series of four peptide crystals using multipole coefficients from the AMOEBA force field demonstrates that AMOEBA systematically underestimates electron density at bond centers. For the trigonal and tetrahedral bonding geometries common in organic chemistry, an atomic multipole expansion through hexadecapole order is required to explain bond electron density. Alternatively, the addition of interatomic scattering (IAS) sites to the AMOEBA-based density captured bonding effects with fewer parameters. For a series of four peptide crystals, the AMOEBA-IAS model lowered R(free) by 20-40% relative to the original spherically symmetric scattering model.
Figures


References
-
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. - PubMed
-
- Afonine, P. V. & Urzhumtsev, A. (2004). Acta Cryst. A60, 19–32. - PubMed
-
- Agarwal, R. C. (1978). Acta Cryst. A34, 791–809.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
