

Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster
- PMID: 19690389
- PMCID: PMC2735940
- DOI: 10.1172/JCI38864
Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster
Abstract
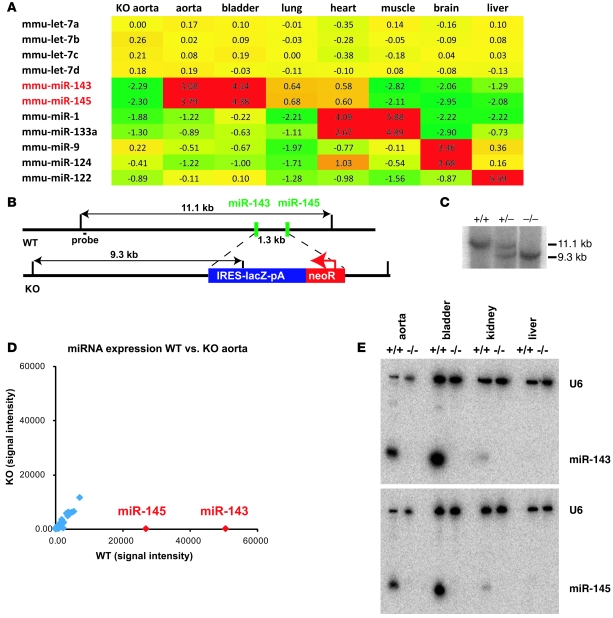
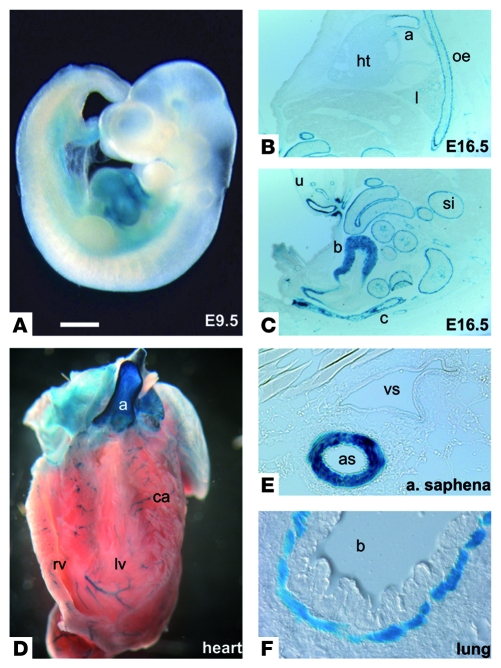
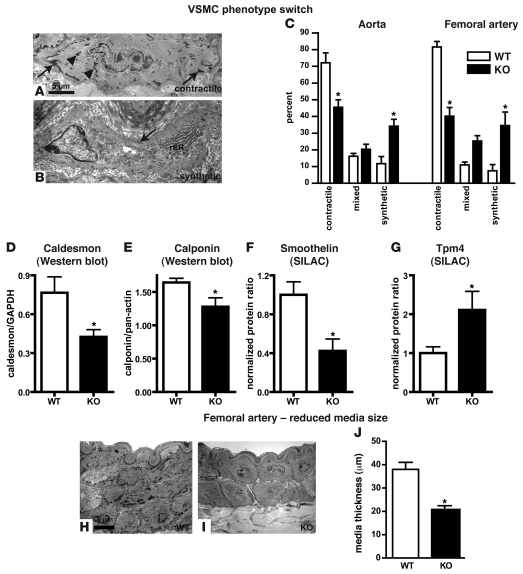
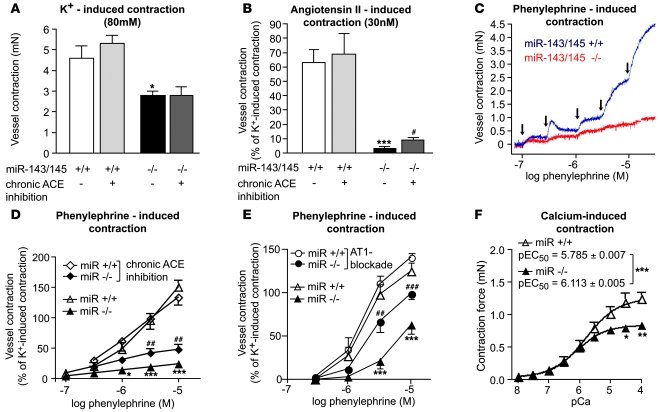
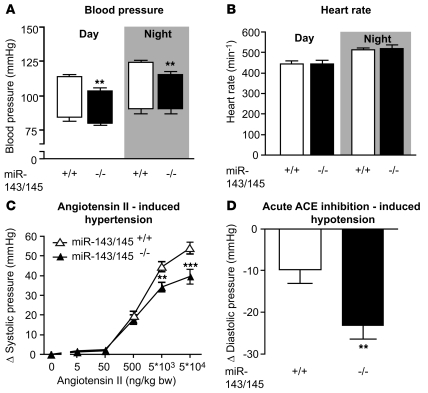
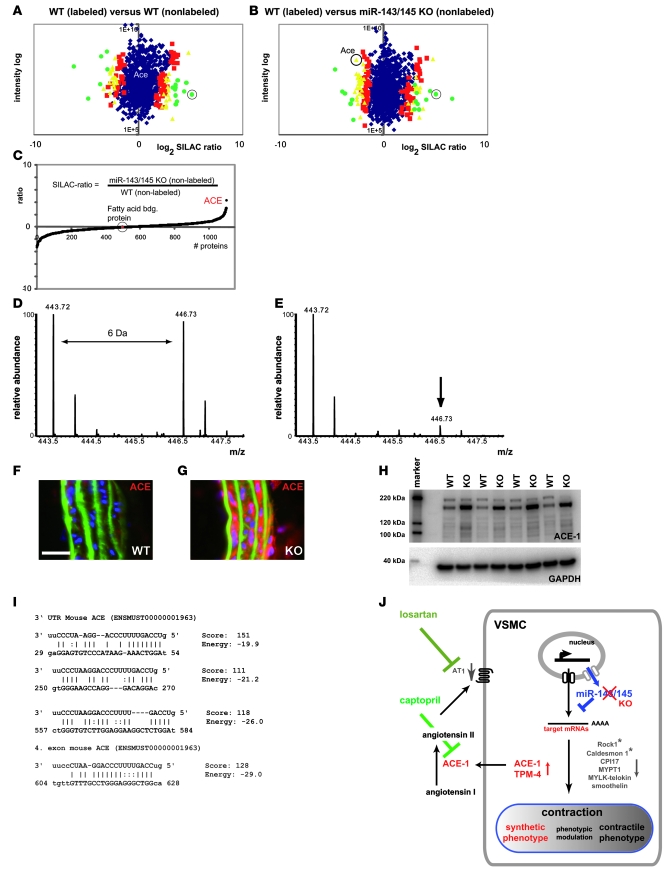
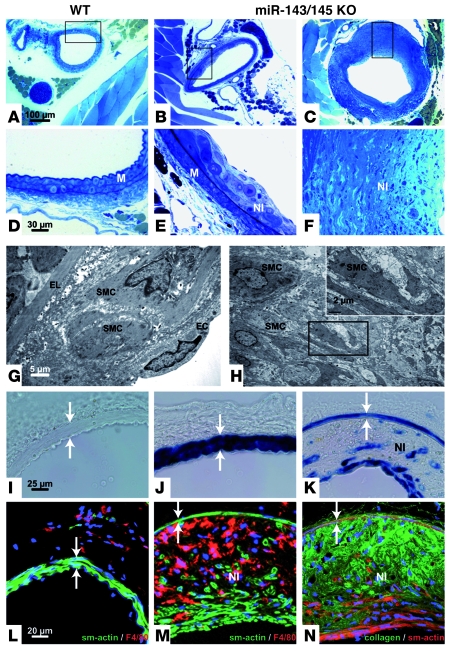
VSMCs respond to changes in the local environment by adjusting their phenotype from contractile to synthetic, a phenomenon known as phenotypic modulation or switching. Failure of VSMCs to acquire and maintain the contractile phenotype plays a key role in a number of major human diseases, including arteriosclerosis. Although several regulatory circuits that control differentiation of SMCs have been identified, the decisive mechanisms that govern phenotypic modulation remain unknown. Here, we demonstrate that the mouse miR-143/145 cluster, expression of which is confined to SMCs during development, is required for VSMC acquisition of the contractile phenotype. VSMCs from miR-143/145-deficient mice were locked in the synthetic state, which incapacitated their contractile abilities and favored neointimal lesion development. Unbiased high-throughput, quantitative, mass spectrometry-based proteomics using reference mice labeled with stable isotopes allowed identification of miR-143/145 targets; these included angiotensin-converting enzyme (ACE), which might affect both the synthetic phenotype and contractile functions of VSMCs. Pharmacological inhibition of either ACE or the AT1 receptor partially reversed vascular dysfunction and normalized gene expression in miR-143/145-deficient mice. We conclude that manipulation of miR-143/145 expression may offer a new approach for influencing vascular repair and attenuating arteriosclerotic pathogenesis.
Figures
Comment in
-
MicroRNA-modulated targeting of vascular smooth muscle cells.J Clin Invest. 2009 Sep;119(9):2526-8. doi: 10.1172/JCI40503. Epub 2009 Aug 17. J Clin Invest. 2009. PMID: 19690387 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
