

The protein meta-structure: a novel concept for chemical and molecular biology
- PMID: 19690801
- PMCID: PMC11115628
- DOI: 10.1007/s00018-009-0117-0
The protein meta-structure: a novel concept for chemical and molecular biology
Abstract
The ultimate goal of bioinformatics or computational chemical biology is the sequence-based prediction of protein functionality. However, due to the degeneracy of the primary sequence code there is no unambiguous relationship. The degeneracy can be partly lifted by going to higher levels of abstraction and, for example, incorporating 3D structural information. However, sometimes even at this conceptual level functional ambiguities often remain. Here a novel conceptual framework is described (the protein meta-structure). At this level of abstraction, the protein structure is viewed as an intricate network of interacting residues. This novel conception offers unique possibilities for chemical (molecular) biology, structural genomics and drug discovery. In this review some prototypical applications will be presented that serve to illustrate the potential of the methodology.
Figures


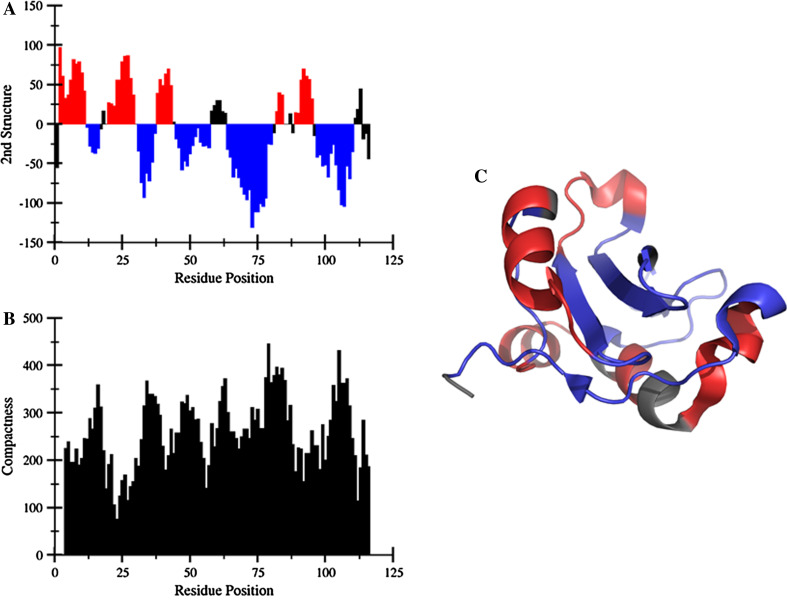


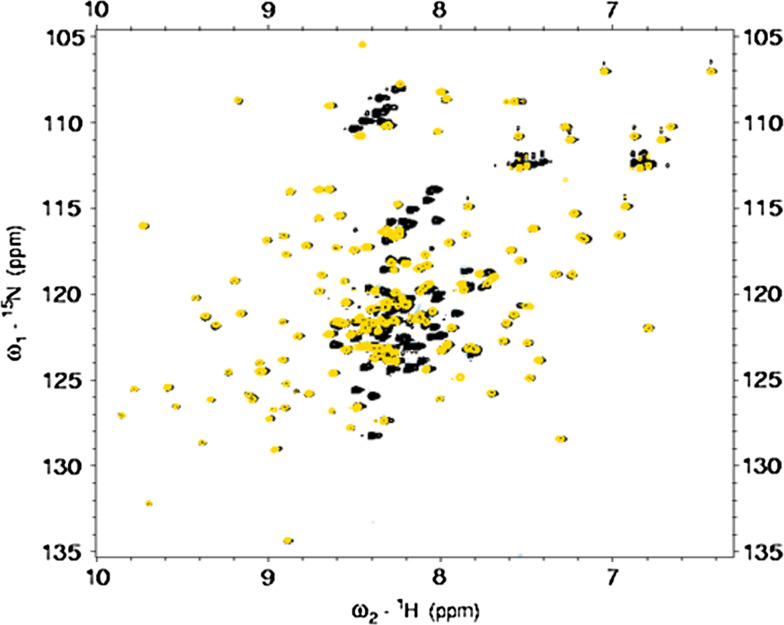
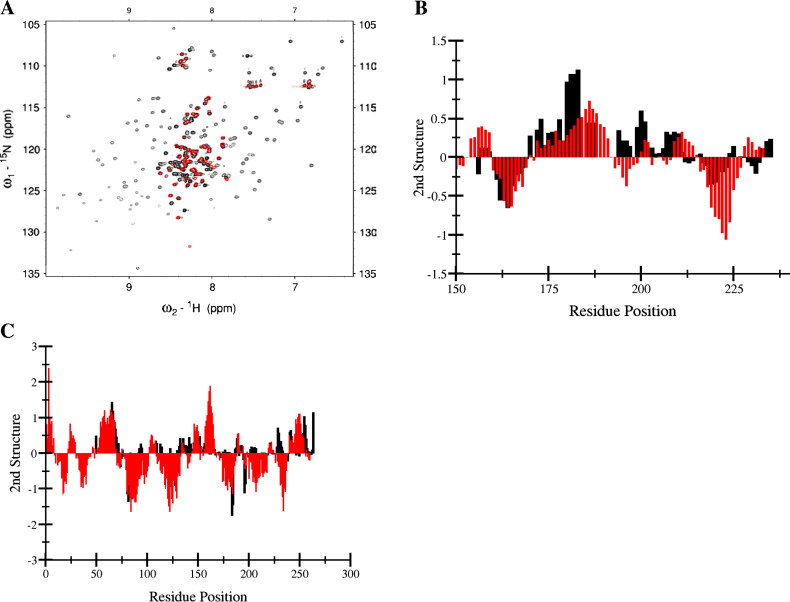
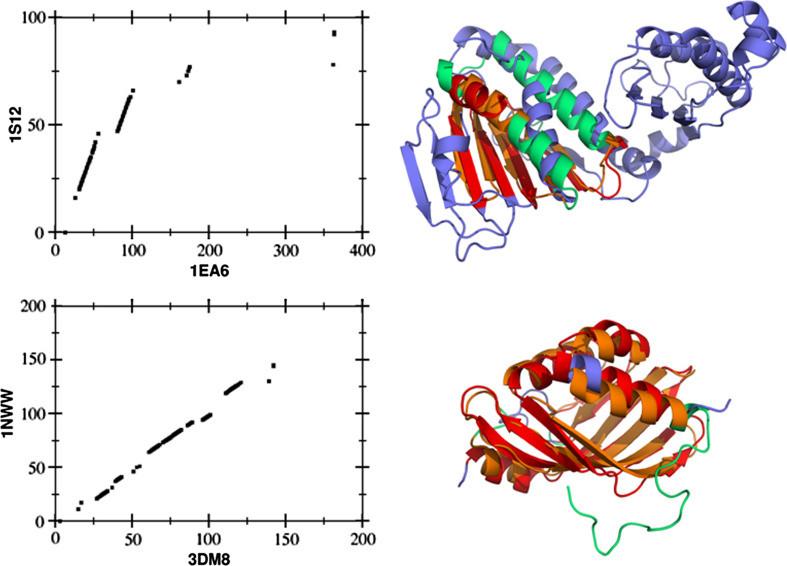
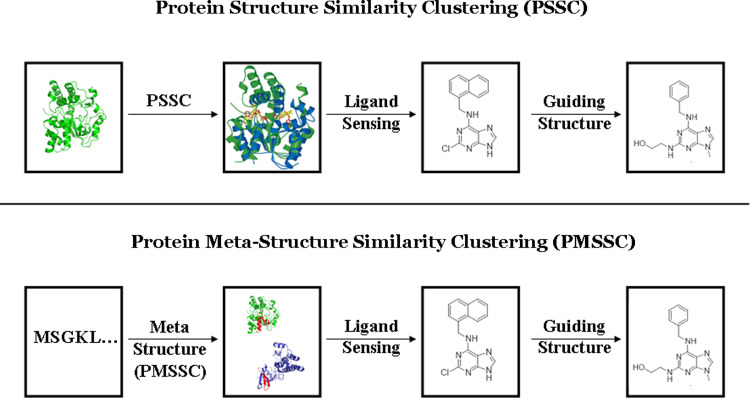
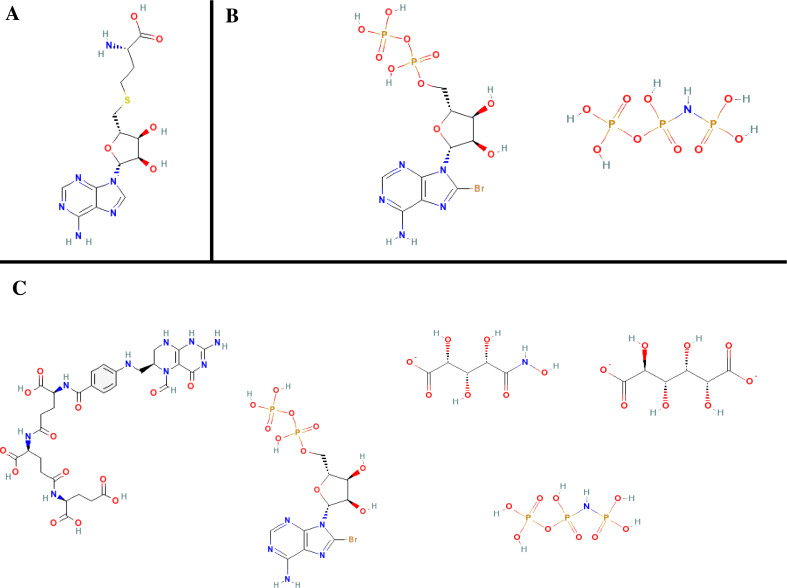
References
-
- Tanford C, Reynolds J (2003) Nature’s robots. A history of proteins. Oxford University Press, Oxford
-
- Epstein CJ, Goldberger RF, Anfinsen CB. The genetic control of tertiary protein structure. Model systems. Cold Spring Harb Symp Quant Biol. 1963;28:439–449.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
