

Heme degradation and vascular injury
- PMID: 19697995
- PMCID: PMC2821146
- DOI: 10.1089/ars.2009.2822
Heme degradation and vascular injury
Abstract
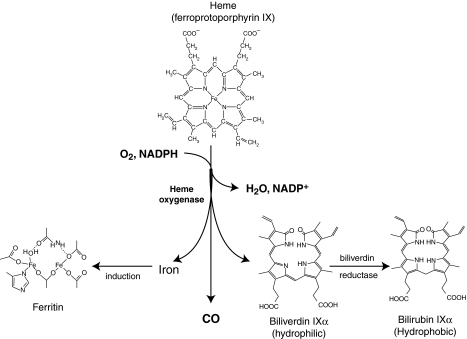
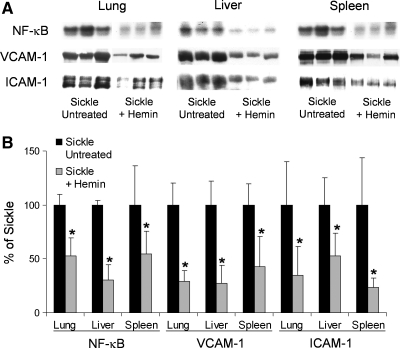
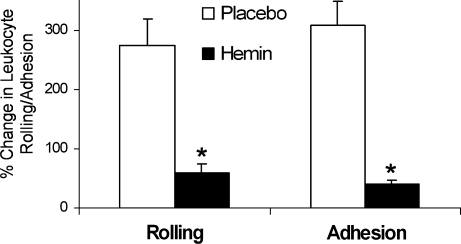
Heme is an essential molecule in aerobic organisms. Heme consists of protoporphyrin IX and a ferrous (Fe(2+)) iron atom, which has high affinity for oxygen (O(2)). Hemoglobin, the major oxygen-carrying protein in blood, is the most abundant heme-protein in animals and humans. Hemoglobin consists of four globin subunits (alpha(2)beta(2)), with each subunit carrying a heme group. Ferrous (Fe(2+)) hemoglobin is easily oxidized in circulation to ferric (Fe(3+)) hemoglobin, which readily releases free hemin. Hemin is hydrophobic and intercalates into cell membranes. Hydrogen peroxide can split the heme ring and release "free" redox-active iron, which catalytically amplifies the production of reactive oxygen species. These oxidants can oxidize lipids, proteins, and DNA; activate cell-signaling pathways and oxidant-sensitive, proinflammatory transcription factors; alter protein expression; perturb membrane channels; and induce apoptosis and cell death. Heme-derived oxidants induce recruitment of leukocytes, platelets, and red blood cells to the vessel wall; oxidize low-density lipoproteins; and consume nitric oxide. Heme metabolism, extracellular and intracellular defenses against heme, and cellular cytoprotective adaptations are emphasized. Sickle cell disease, an archetypal example of hemolysis, heme-induced oxidative stress, and cytoprotective adaptation, is reviewed.
Figures







References
-
- Agarwal A. Kim Y. Matas A. Alam J. Nath K. Gas-generation systems in acute renal allograft rejection in the rat: co-Induction of heme oxygenase and nitric oxide synthase. Transplantation. 1996;61:93–98. - PubMed
-
- Akerström B. Tissue distribution of guinea pig alpha 1-microglobulin. Cell Mol Biol. 1983;29:489–495. - PubMed
-
- Alam J. Multiple elements within the 5′ distal enhancer of the mouse heme oxygenase-1 gene mediate induction by heavy metals. J Biol Chem. 1994;269:25049–25056. - PubMed
-
- Alam J. Camhi S. Choi AMK. Identification of a second region upstream of the mouse heme oxygenase-1 gene that functions as a basal level and inducer-dependent transcription enhancer. J Biol Chem. 1995;270:11977–11984. - PubMed
-
- Alam J. Den Z. Distal AP-1 binding sites mediate basal level enhancement and TPA induction of the mouse heme oxygenase-1 gene. J Biol Chem. 1992;267:21894–21900. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
