

Transfusion-related acute lung injury (TRALI): current concepts and misconceptions
- PMID: 19699017
- PMCID: PMC3134874
- DOI: 10.1016/j.blre.2009.07.005
Transfusion-related acute lung injury (TRALI): current concepts and misconceptions
Abstract
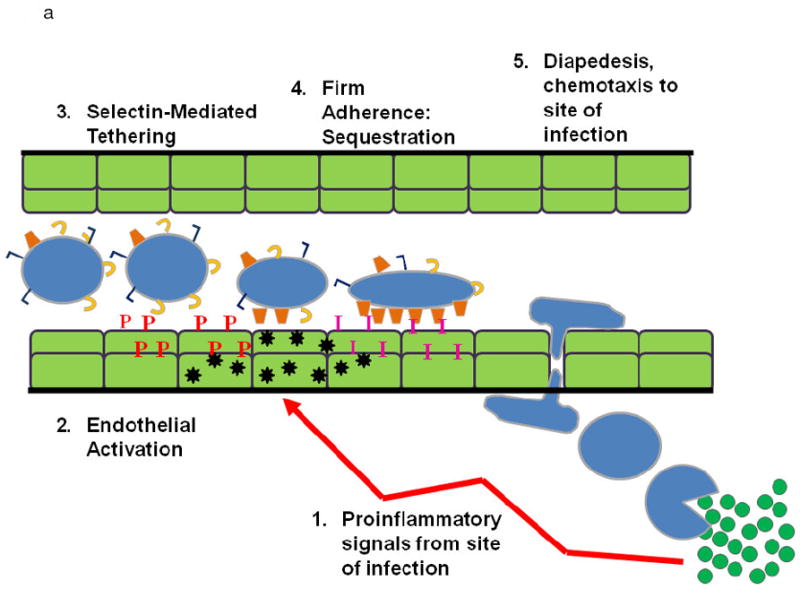
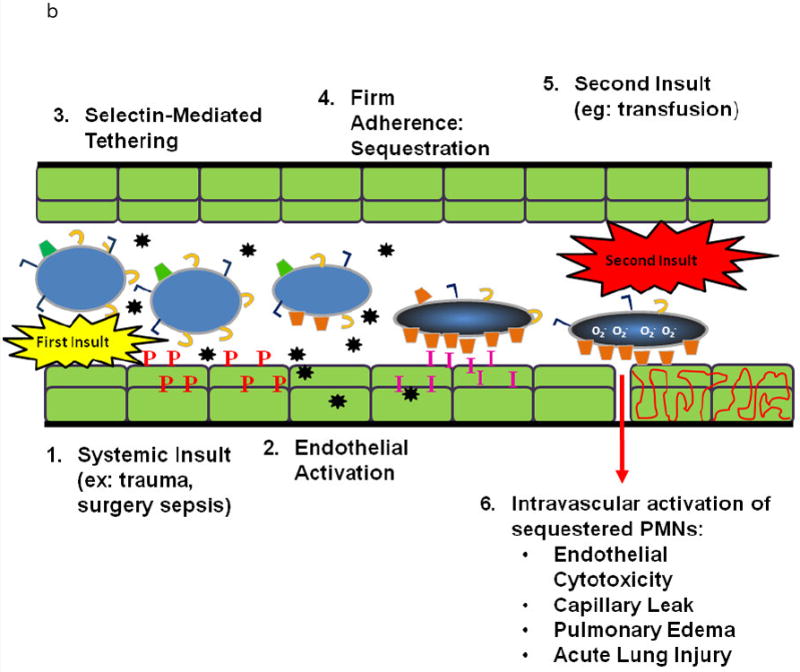
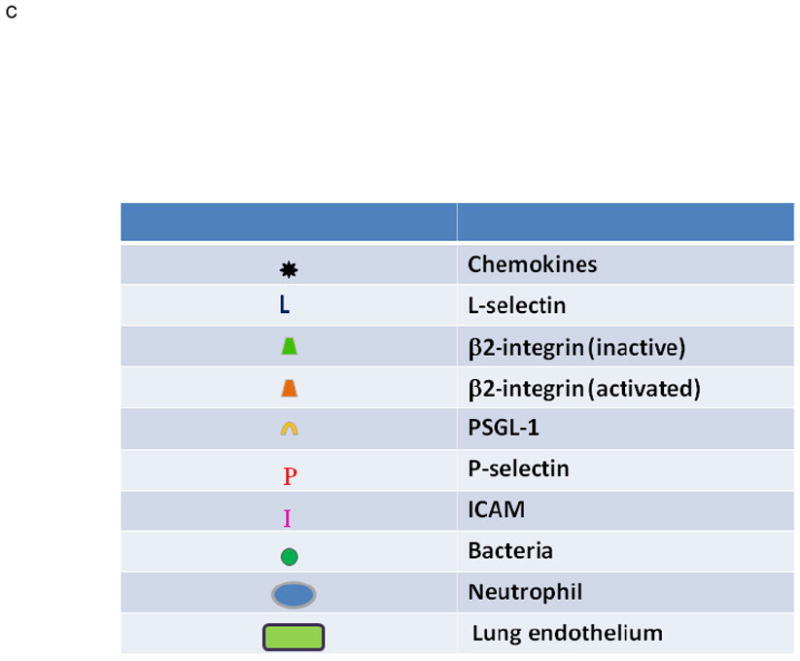
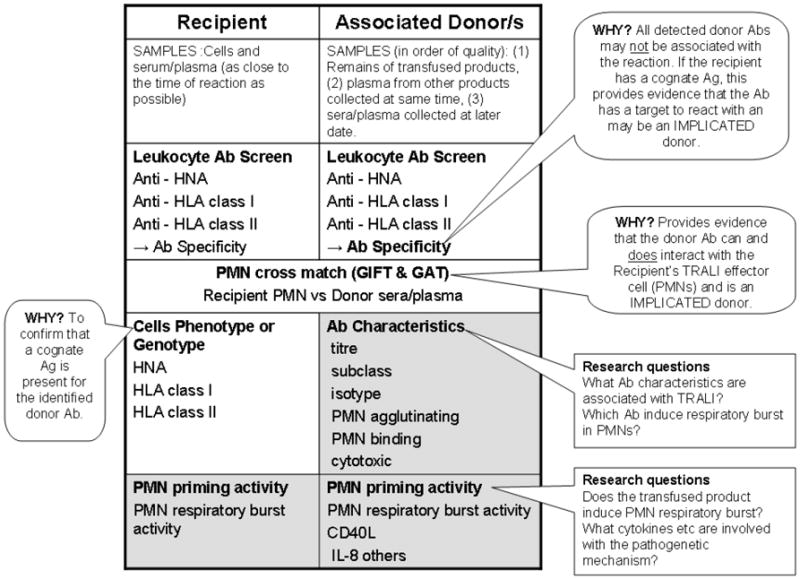
Transfusion-related acute lung injury (TRALI) is the most common cause of serious morbidity and mortality due to hemotherapy. Although the pathogenesis has been related to the infusion of donor antibodies into the recipient, antibody negative TRALI has been reported. Changes in transfusion practices, especially the use of male-only plasma, have decreased the number of antibody-mediated cases and deaths; however, TRALI still occurs. The neutrophil appears to be the effector cell in TRALI and the pathophysiology is centered on neutrophil-mediated endothelial cell cytotoxicity resulting in capillary leak and ALI. This review will detail the pathophysiology of TRALI including recent pre-clinical data, provide insight into newer areas of research, and critically assess current practices to decrease it prevalence and to make transfusion safer.
Figures
References
-
- Popovsky MA, Moore SB. Diagnostic and pathogenetic considerations in transfusion-related acute lung injury. Transfusion. 1985 November;25(6):573–7. - PubMed
-
- Kleinman S, Caulfield T, Chan P, Davenport R, McFarland J, McPhedran S, Meade M, Morrison D, Pinsent T, Robillard P, Slinger P. Toward an understanding of transfusion-related acute lung injury: statement of a consensus panel. Transfusion. 2004 December;44(12):1774–89. - PubMed
-
- Toy P, Popovsky MA, Abraham E, Ambruso DR, Holness LG, Kopko PM, McFarland JG, Nathens AB, Silliman CC, Stroncek D. Transfusion-related acute lung injury: definition and review. Crit Care Med. 2005 April;33(4):721–6. - PubMed
-
- Silliman CC, Paterson AJ, Dickey WO, Stroneck DF, Popovsky MA, Caldwell SA, Ambruso DR. The association of biologically active lipids with the development of transfusion-related acute lung injury: a retrospective study. Transfusion. 1997 July;37(7):719–26. - PubMed
-
- Silliman CC, Boshkov LK, Mehdizadehkashi Z, Elzi DJ, Dickey WO, Podlosky L, Clarke G, Ambruso DR. Transfusion-related acute lung injury: epidemiology and a prospective analysis of etiologic factors. Blood. 2003 January 15;101(2):454–62. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
