

Convergent Random Forest predictor: methodology for predicting drug response from genome-scale data applied to anti-TNF response
- PMID: 19699293
- PMCID: PMC4476397
- DOI: 10.1016/j.ygeno.2009.08.008
Convergent Random Forest predictor: methodology for predicting drug response from genome-scale data applied to anti-TNF response
Abstract
Biomarker development for prediction of patient response to therapy is one of the goals of molecular profiling of human tissues. Due to the large number of transcripts, relatively limited number of samples, and high variability of data, identification of predictive biomarkers is a challenge for data analysis. Furthermore, many genes may be responsible for drug response differences, but often only a few are sufficient for accurate prediction. Here we present an analysis approach, the Convergent Random Forest (CRF) method, for the identification of highly predictive biomarkers. The aim is to select from genome-wide expression data a small number of non-redundant biomarkers that could be developed into a simple and robust diagnostic tool. Our method combines the Random Forest classifier and gene expression clustering to rank and select a small number of predictive genes. We evaluated the CRF approach by analyzing four different data sets. The first set contains transcript profiles of whole blood from rheumatoid arthritis patients, collected before anti-TNF treatment, and their subsequent response to the therapy. In this set, CRF identified 8 transcripts predicting response to therapy with 89% accuracy. We also applied the CRF to the analysis of three previously published expression data sets. For all sets, we have compared the CRF and recursive support vector machines (RSVM) approaches to feature selection and classification. In all cases the CRF selects much smaller number of features, five to eight genes, while achieving similar or better performance on both training and independent testing sets of data. For both methods performance estimates using cross-validation is similar to performance on independent samples. The method has been implemented in R and is available from the authors upon request: Jadwiga.Bienkowska@biogenidec.com.
Figures
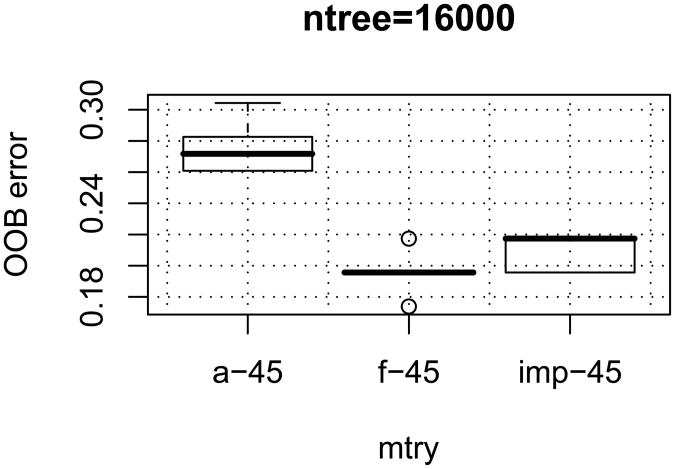
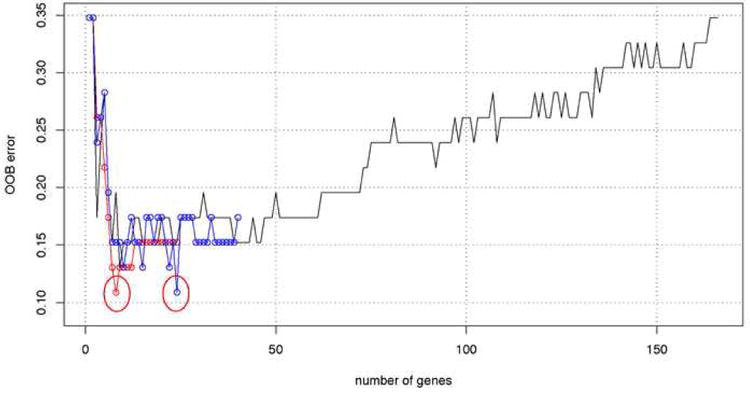
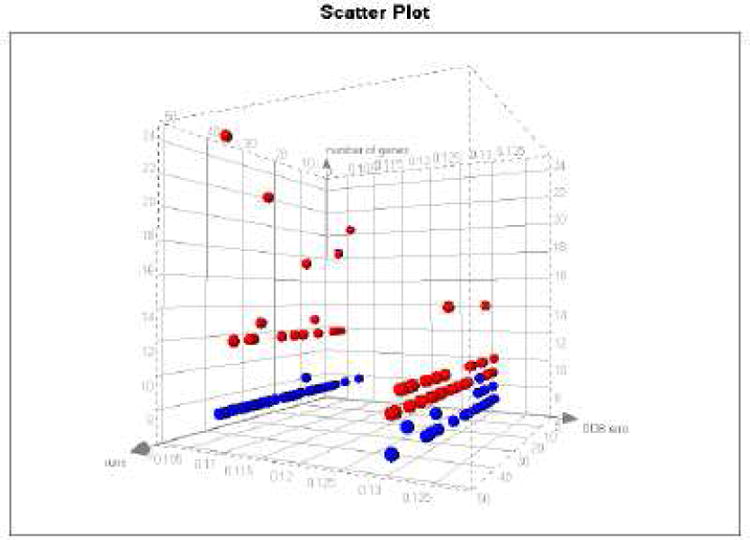
References
-
- Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Machine Learning. 2002;46:389–422.
-
- Krishnapuram B, Carin L, Hartemink AJ. Joint classifier and feature optimization for comprehensive cancer diagnosis using gene expression data. Journal of Computational Biology. 2004;11:227–42. - PubMed
-
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7. - PubMed
-
- Breiman L. Random forests. Machine Learning. 2001;45:5–32.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
