

Structure prediction for CASP8 with all-atom refinement using Rosetta
- PMID: 19701941
- PMCID: PMC3688471
- DOI: 10.1002/prot.22540
Structure prediction for CASP8 with all-atom refinement using Rosetta
Abstract
We describe predictions made using the Rosetta structure prediction methodology for the Eighth Critical Assessment of Techniques for Protein Structure Prediction. Aggressive sampling and all-atom refinement were carried out for nearly all targets. A combination of alignment methodologies was used to generate starting models from a range of templates, and the models were then subjected to Rosetta all atom refinement. For the 64 domains with readily identified templates, the best submitted model was better than the best alignment to the best template in the Protein Data Bank for 24 cases, and improved over the best starting model for 43 cases. For 13 targets where only very distant sequence relationships to proteins of known structure were detected, models were generated using the Rosetta de novo structure prediction methodology followed by all-atom refinement; in several cases the submitted models were better than those based on the available templates. Of the 12 refinement challenges, the best submitted model improved on the starting model in seven cases. These improvements over the starting template-based models and refinement tests demonstrate the power of Rosetta structure refinement in improving model accuracy.
Copyright 2009 Wiley-Liss, Inc.
Figures
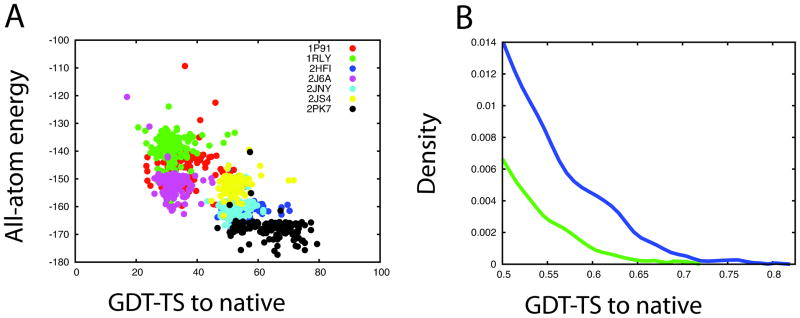
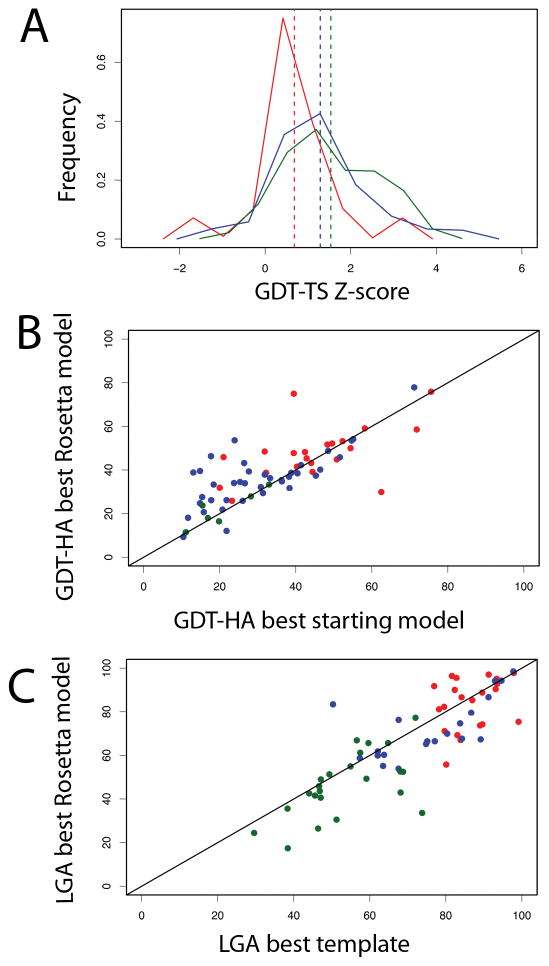

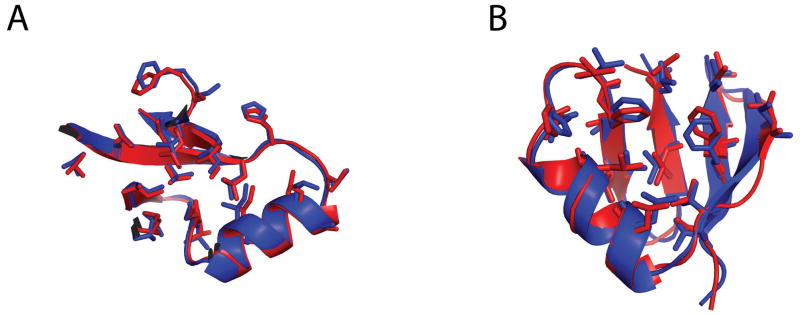


References
-
- Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. - PubMed
-
- Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309(5742):1868–1871. - PubMed
-
- Baker D, Sohl JL, Agard DA. A protein-folding reaction under kinetic control. Nature. 1992;356(6366):263–265. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
