

Protein sectors: evolutionary units of three-dimensional structure
- PMID: 19703402
- PMCID: PMC3210731
- DOI: 10.1016/j.cell.2009.07.038
Protein sectors: evolutionary units of three-dimensional structure
Abstract
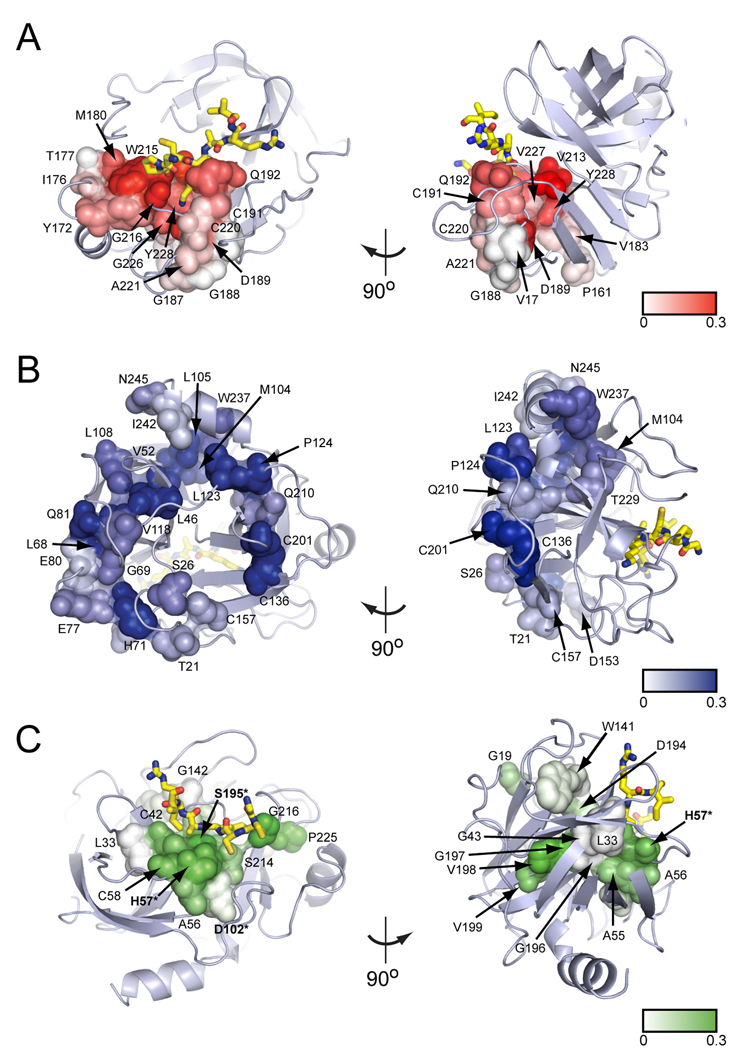
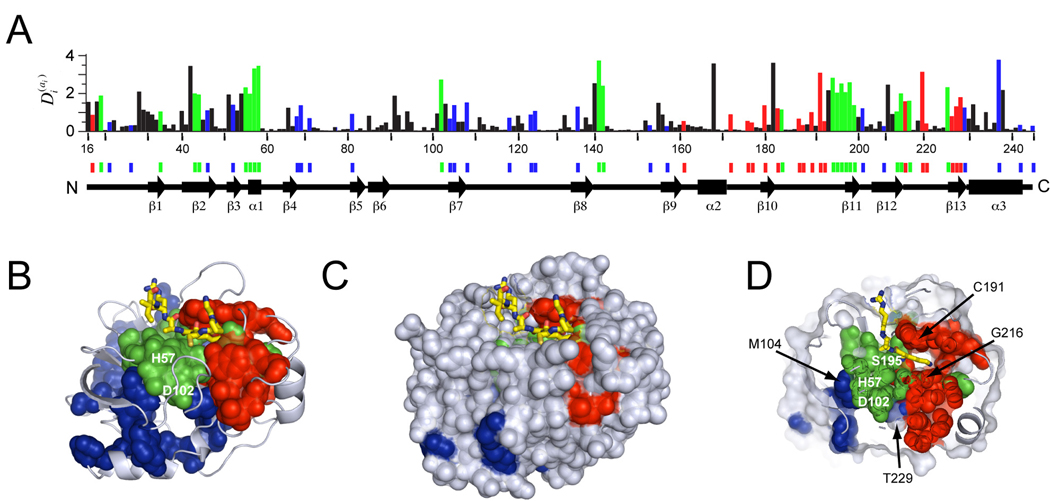
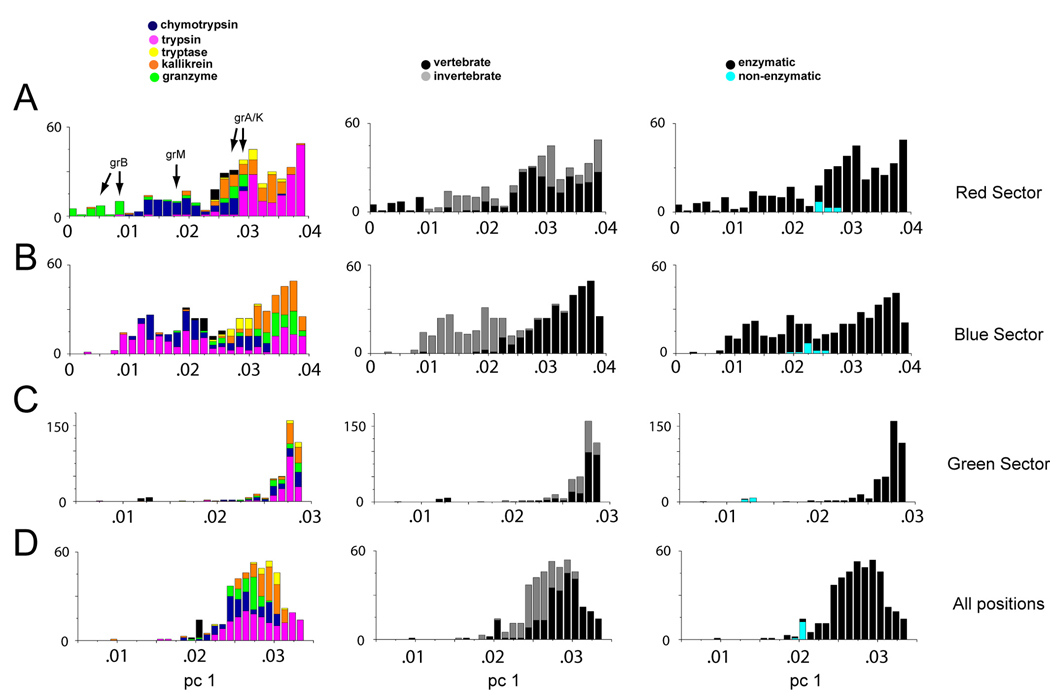
Proteins display a hierarchy of structural features at primary, secondary, tertiary, and higher-order levels, an organization that guides our current understanding of their biological properties and evolutionary origins. Here, we reveal a structural organization distinct from this traditional hierarchy by statistical analysis of correlated evolution between amino acids. Applied to the S1A serine proteases, the analysis indicates a decomposition of the protein into three quasi-independent groups of correlated amino acids that we term "protein sectors." Each sector is physically connected in the tertiary structure, has a distinct functional role, and constitutes an independent mode of sequence divergence in the protein family. Functionally relevant sectors are evident in other protein families as well, suggesting that they may be general features of proteins. We propose that sectors represent a structural organization of proteins that reflects their evolutionary histories.
Figures




References
-
- Atchley WR, Wollenberg KR, Fitch WM, Terhalle W, Dress AW. Correlations among amino acid sites in bHLH protein domains: an information theoretic analysis. Mol Biol Evol. 2000;17:164–178. - PubMed
-
- Bell JK, Goetz DH, Mahrus S, Harris JL, Fletterick RJ, Craik CS. The oligomeric structure of human granzyme A is a determinant of its extended substrate specificity. Nat Struct Biol. 2003;10:527–534. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
