

The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism
- PMID: 19707180
- PMCID: PMC2864719
- DOI: 10.1038/nrendo.2009.177
The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism
Abstract
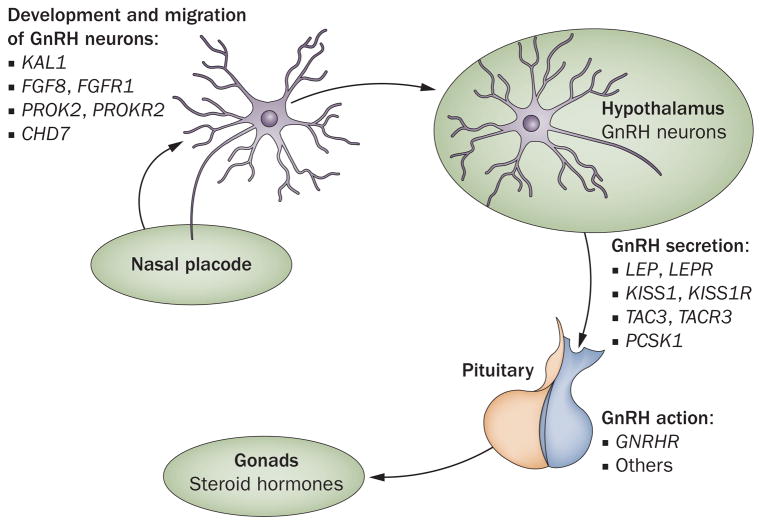
Idiopathic hypogonadotropic hypogonadism (IHH) has an incidence of 1-10 cases per 100,000 births. About 60% of patients with IHH present with associated anosmia, also known as Kallmann syndrome, characterized by total or partial loss of olfaction. Many of the gene mutations associated with Kallmann syndrome have been mapped to KAL1 or FGFR1. However, together, these mutations account for only about 15% of Kallmann syndrome cases. More recently, mutations in PROK2 and PROKR2 have been linked to the syndrome and may account for an additional 5-10% of cases. The remaining 40% of patients with IHH have a normal sense of smell. Prior to 2003, the only gene linked to normosmic IHH was the gonadotropin-releasing hormone receptor gene. However, mutations in this receptor are believed to account for only 10% of cases. Subsequently, mutations in KISS1R, TAC3 and TACR3 were identified as causes of normosmic IHH. Certain genes, including PROK2 and FGFR1, are associated with both anosmic and normosmic IHH. Despite recent advances in the field, the genetic causes of the majority of cases of IHH remain unknown. This Review discusses genes associated with hypogonadotropic disorders and the molecular mechanisms by which mutations in these genes may result in IHH.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Cadman SM, Kim SH, Hu Y, González-Martínez D, Bouloux PM. Molecular pathogenesis of Kallmann’s syndrome. Horm Res. 2007;67:231–242. - PubMed
-
- Schwanzel-Fukuda M. Origin and migration of luteinizing hormone-releasing hormone neurons in mammals. Microsc Res Tech. 1999;44:2–10. - PubMed
-
- Pitteloud N, et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. 2006;254–255:60–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
