

PAVE: program for assembling and viewing ESTs
- PMID: 19709403
- PMCID: PMC2748094
- DOI: 10.1186/1471-2164-10-400
PAVE: program for assembling and viewing ESTs
Abstract
Background: New sequencing technologies are rapidly emerging. Many laboratories are simultaneously working with the traditional Sanger ESTs and experimenting with ESTs generated by the 454 Life Science sequencers. Though Sanger ESTs have been used to generate contigs for many years, no program takes full advantage of the 5' and 3' mate-pair information, hence, many tentative transcripts are assembled into two separate contigs. The new 454 technology has the benefit of high-throughput expression profiling, but introduces time and space problems for assembling large contigs.
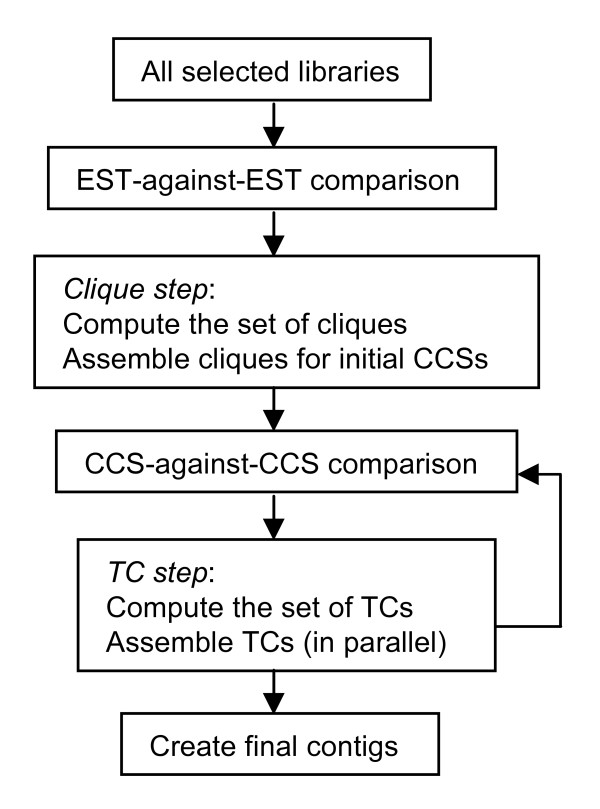
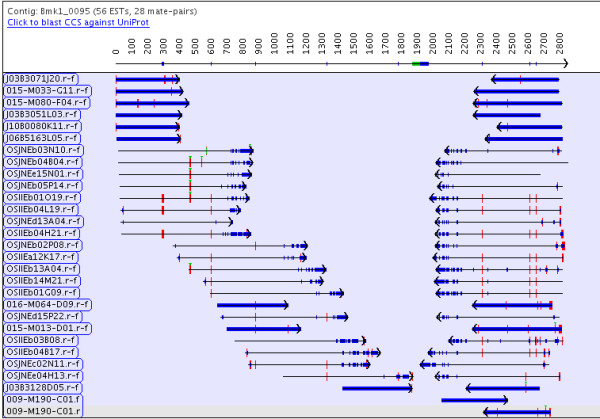
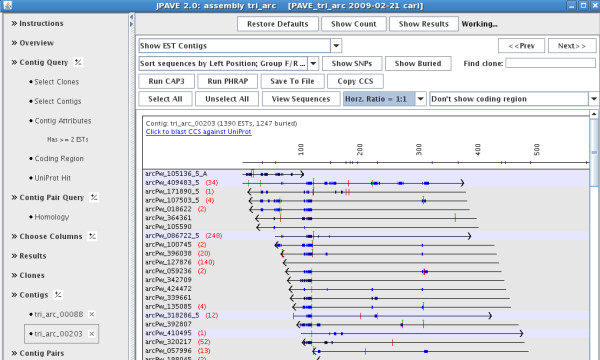
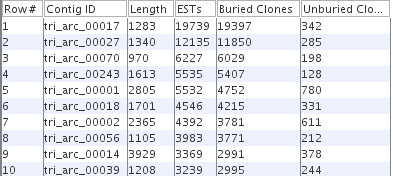
Results: The PAVE (Program for Assembling and Viewing ESTs) assembler takes advantage of the 5' and 3' mate-pair information by requiring that the mate-pairs be assembled into the same contig and joined by n's if the two sub-contigs do not overlap. It handles the depth of 454 data sets by "burying" similar ESTs during assembly, which retains the expression level information while circumventing time and space problems. PAVE uses MegaBLAST for the clustering step and CAP3 for assembly, however it assembles incrementally to enforce the mate-pair constraint, bury ESTs, and reduce incorrect joins and splits. The PAVE data management system uses a MySQL database to store multiple libraries of ESTs along with their metadata; the management system allows multiple assemblies with variations on libraries and parameters. Analysis routines provide standard annotation for the contigs including a measure of differentially expressed genes across the libraries. A Java viewer program is provided for display and analysis of the results. Our results clearly show the benefit of using the PAVE assembler to explicitly use mate-pair information and bury ESTs for large contigs.
Conclusion: The PAVE assembler provides a software package for assembling Sanger and/or 454 ESTs. The assembly software, data management software, Java viewer and user's guide are freely available.
Figures
References
-
- 454 Life Sciences, a Roche Company http://www.454.com
-
- Solexa/Illumina http://www.illumina.com
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
