

The ERCC1/XPF endonuclease is required for completion of homologous recombination at DNA replication forks stalled by inter-strand cross-links
- PMID: 19713438
- PMCID: PMC2770670
- DOI: 10.1093/nar/gkp705
The ERCC1/XPF endonuclease is required for completion of homologous recombination at DNA replication forks stalled by inter-strand cross-links
Abstract
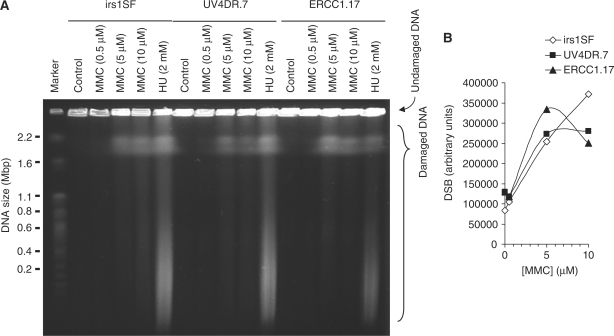
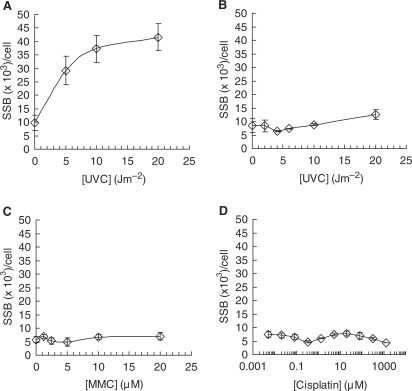
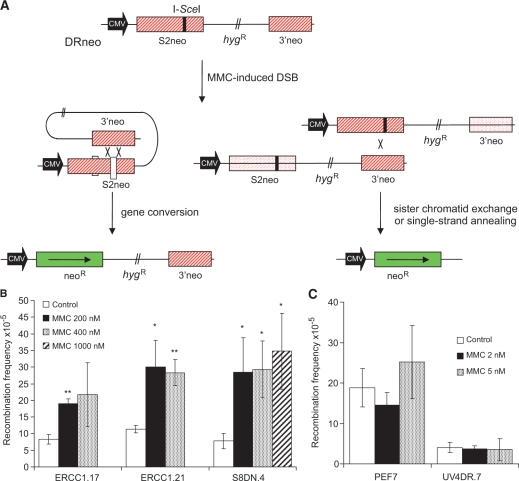
Both the ERCC1-XPF complex and the proteins involved in homoIogous recombination (HR) have critical roles in inter-strand cross-link (ICL) repair. Here, we report that mitomycin C-induced lesions inhibit replication fork elongation. Furthermore, mitomycin C-induced DNA double-strand breaks (DSBs) are the result of the collapse of ICL-stalled replication forks. These are not formed through replication run off, as we show that mitomycin C or cisplatin-induced DNA lesions are not incised by global genome nucleotide excision repair (GGR). We also suggest that ICL-lesion repair is initiated either by replication or transcription, as the GGR does not incise ICL-lesions. Furthermore, we report that RAD51 foci are induced by cisplatin or mitomycin C independently of ERCC1, but that mitomycin C-induced HR measured in a reporter construct is impaired in ERCC1-defective cells. These data suggest that ERCC1-XPF plays a role in completion of HR in ICL repair. We also find no additional sensitivity to cisplatin by siRNA co-depletion of XRCC3 and ERCC1, showing that the two proteins act on the same pathway to promote survival.
Figures







References
-
- Lawley PD, Phillips DH. DNA adducts from chemotherapeutic agents. Mutat. Res. 1996;355:13–40. - PubMed
-
- Palom Y, Suresh Kumar G, Tang LQ, Paz MM, Musser SM, Rockwell S, Tomasz M. Relative toxicities of DNA cross-links and monoadducts: new insights from studies of decarbamoyl mitomycin C and mitomycin C. Chem. Res. Toxicol. 2002;15:1398–1406. - PubMed
-
- Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat. Res. 2001;486:217–247. - PubMed
-
- McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2:483–490. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
