

Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression
- PMID: 19713933
- PMCID: PMC2771084
- DOI: 10.1038/emboj.2009.246
Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression
Abstract
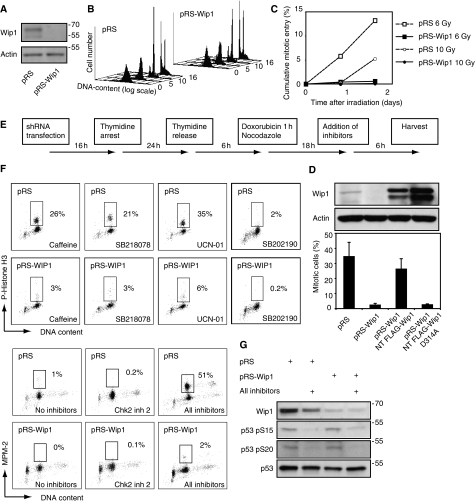
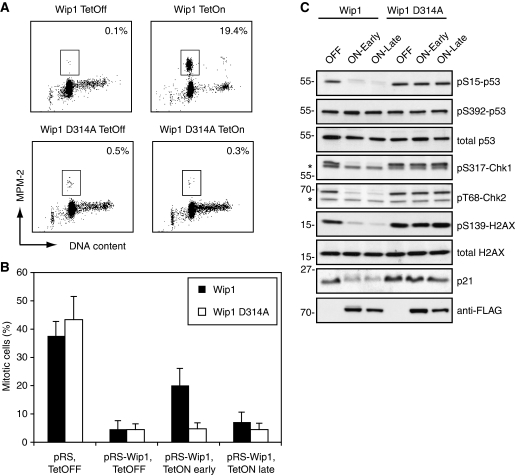
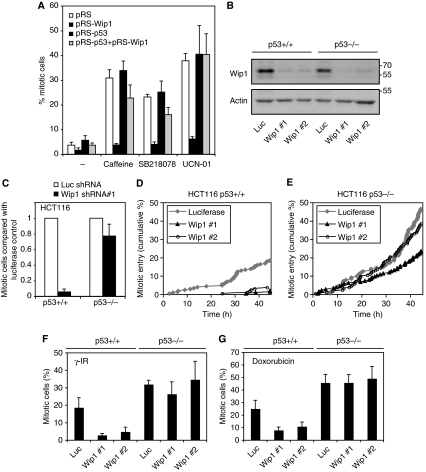
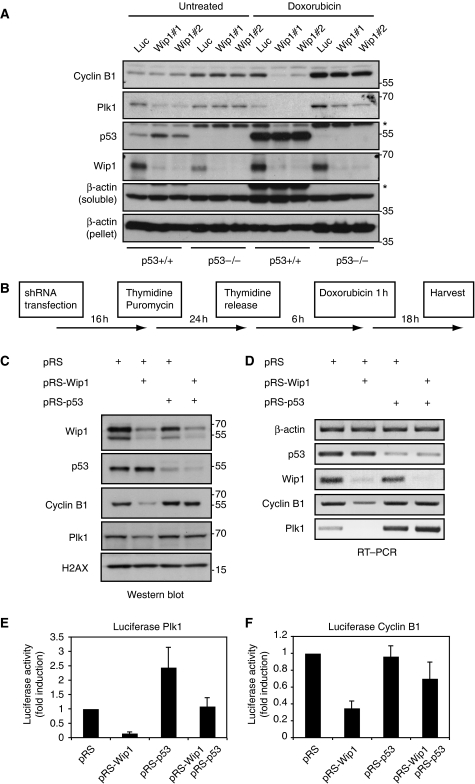
Activation of the DNA damage checkpoint causes a cell-cycle arrest through inhibition of cyclin-dependent kinases (cdks). To successfully recover from the arrest, a cell should somehow be maintained in its proper cell-cycle phase. This problem is particularly eminent when a cell arrests in G2, as cdk activity is important to establish a G2 state. Here, we identify the phosphatase Wip1 (PPM1D) as a factor that maintains a cell competent for cell-cycle re-entry during an ongoing DNA damage response in G2. We show that Wip1 function is required throughout the arrest, and that Wip1 acts by antagonizing p53-dependent repression of crucial mitotic inducers, such as Cyclin B and Plk1. Our data show that the primary function of Wip1 is to retain cellular competence to divide, rather than to silence the checkpoint to promote recovery. Our findings uncover Wip1 as a first in class recovery competence gene, and suggest that the principal function of Wip1 in cellular transformation is to retain proliferative capacity in the face of oncogene-induced stress.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Appella E, Anderson CW (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 268: 2764–2772 - PubMed
-
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281: 1674–1677 - PubMed
-
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LVF, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J et al. (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633–637 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
