

Macrophage derived chemokine (CCL22), thymus and activation-regulated chemokine (CCL17), and CCR4 in idiopathic pulmonary fibrosis
- PMID: 19715610
- PMCID: PMC2741459
- DOI: 10.1186/1465-9921-10-80
Macrophage derived chemokine (CCL22), thymus and activation-regulated chemokine (CCL17), and CCR4 in idiopathic pulmonary fibrosis
Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is a chronically progressive interstitial lung disease of unknown etiology. Previously, we have demonstrated the selective upregulation of the macrophage-derived chemokine CCL22 and the thymus activation-regulated chemokine CCL17 among chemokines, in a rat model of radiation pneumonitis/pulmonary fibrosis and preliminarily observed an increase in bronchoalveolar (BAL) fluid CCL22 levels of IPF patients.
Methods: We examined the expression of CCR4, a specific receptor for CCL22 and CCL17, in bronchoalveolar lavage (BAL) fluid cells, as well as the levels of CCL22 and CCL17, to elucidate their pathophysiological roles in pulmonary fibrosis. We also studied their immunohistochemical localization.
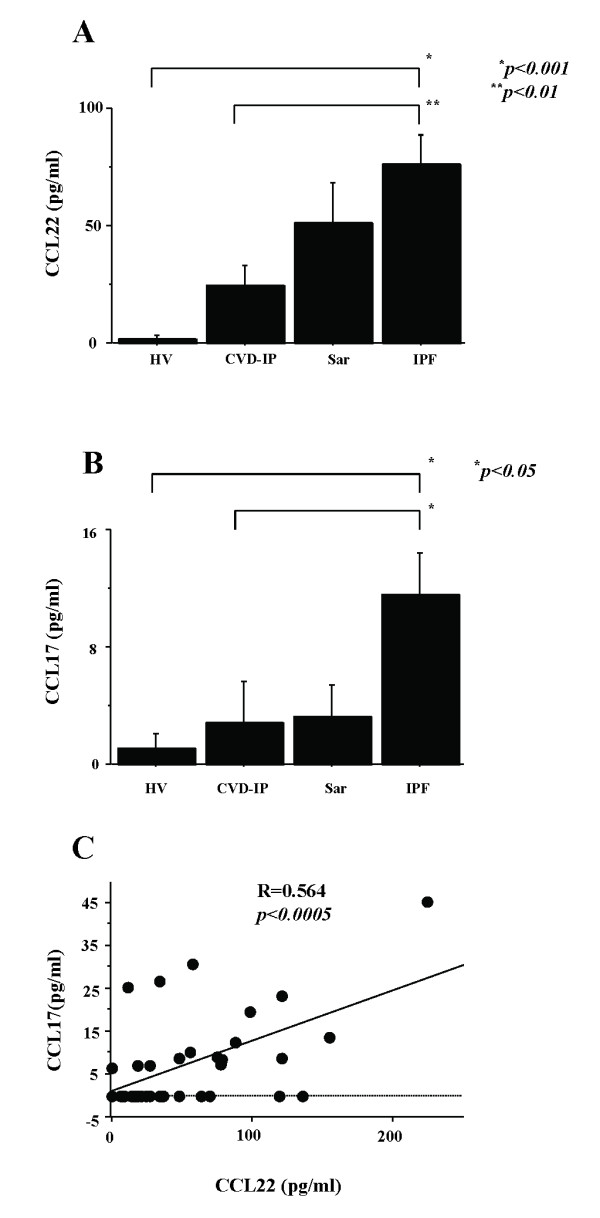
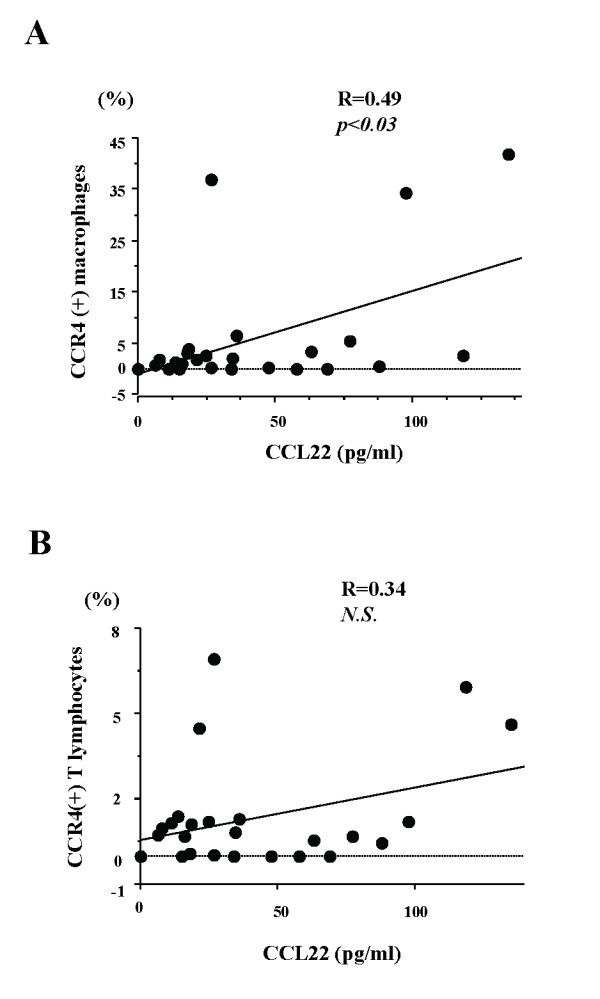
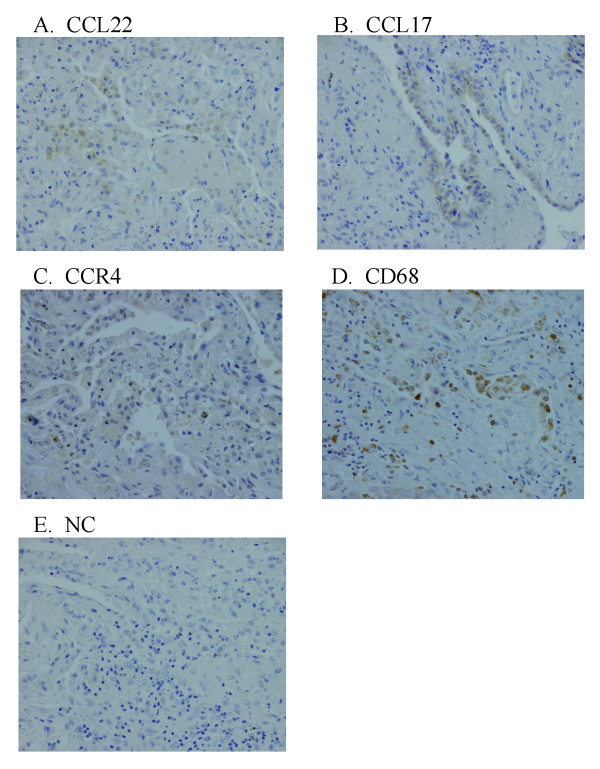
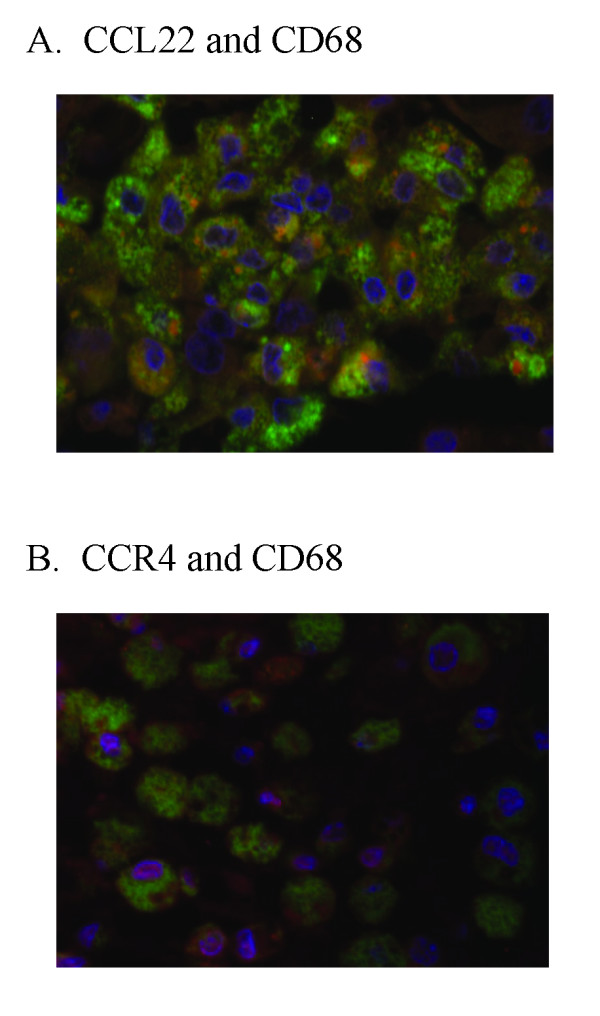
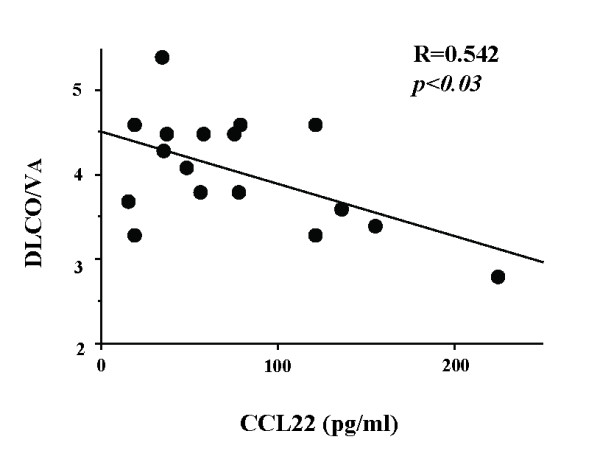
Results: BAL fluid CCL22 and CCL17 levels were significantly higher in patients with IPF than those with collagen vascular diseases and healthy volunteers, and there was a significant correlation between the levels of CCL22 and CCL17 in patients with IPF. CCL22 levels in the BAL fluid did not correlate with the total cell numbers, alveolar lymphocytes, or macrophages in BAL fluid. However, the CCL22 levels significantly correlated with the numbers of CCR4-expressing alveolar macrophages. By immunohistochemical and immunofluorescence analysis, localization of CCL22 and CCR4 to CD68-positive alveolar macrophages as well as that of CCL17 to hyperplastic epithelial cells were shown. Clinically, CCL22 BAL fluid levels inversely correlated with DLco/VA values in IPF patients.
Conclusion: We speculated that locally overexpressed CCL22 may induce lung dysfunction through recruitment and activation of CCR4-positive alveolar macrophages.
Figures
References
-
- American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165(2):277–304. - PubMed
-
- American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161(2 Pt 1):646–664. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
