

Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test
- PMID: 19716085
- PMCID: PMC3049907
- DOI: 10.1016/j.hrthm.2009.05.021
Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test
Abstract
Background: Long QT syndrome (LQTS) is a potentially lethal, highly treatable cardiac channelopathy for which genetic testing has matured from discovery to translation and now clinical implementation.
Objectives: Here we examine the spectrum and prevalence of mutations found in the first 2,500 unrelated cases referred for the FAMILION LQTS clinical genetic test.
Methods: Retrospective analysis of the first 2,500 cases (1,515 female patients, average age at testing 23 +/- 17 years, range 0 to 90 years) scanned for mutations in 5 of the LQTS-susceptibility genes: KCNQ1 (LQT1), KCNH2 (LQT2), SCN5A (LQT3), KCNE1 (LQT5), and KCNE2 (LQT6).
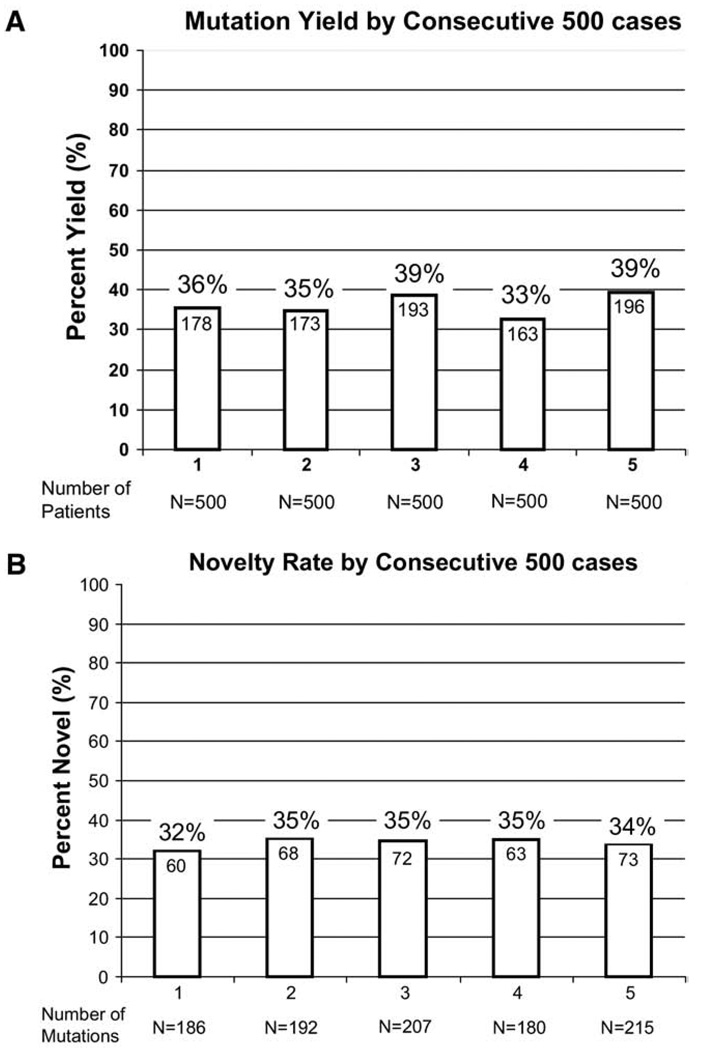
Results: Overall, 903 referral cases (36%) hosted a possible LQTS-causing mutation that was absent in >2,600 reference alleles; 821 (91%) of the mutation-positive cases had single genotypes, whereas the remaining 82 patients (9%) had >1 mutation in > or =1 gene, including 52 cases that were compound heterozygous with mutations in >1 gene. Of the 562 distinct mutations, 394 (70%) were missense, 428 (76%) were seen once, and 336 (60%) are novel, including 92 of 199 in KCNQ1, 159 of 226 in KCNH2, and 70 of 110 in SCN5A.
Conclusion: This cohort increases the publicly available compendium of putative LQTS-associated mutations by >50%, and approximately one-third of the most recently detected mutations continue to be novel. Although control population data suggest that the great majority of these mutations are pathogenic, expert interpretation of genetic test results will remain critical for effective clinical use of LQTS genetic test results.
Figures



Comment in
-
Is it time to develop a "pathogenicity" score to distinguish long QT syndrome causing mutations from "background" genetic noise?Heart Rhythm. 2009 Sep;6(9):1304-5. doi: 10.1016/j.hrthm.2009.06.027. Epub 2009 Jun 25. Heart Rhythm. 2009. PMID: 19716086 Free PMC article. No abstract available.
References
-
- Ackerman MJ. Cardiac channelopathies: it’s in the genes. Nat Med. 2004;10:463–464. - PubMed
-
- Stramba-Badiale M, Crotti L, Goulene K, et al. Electrocardiographic and genetic screening for long QT syndrome: results from a prospective study on 44,596 neonates. Circulation. 2007;116 (abstract 1778)II_377.
-
- Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. - PubMed
-
- Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. - PubMed
-
- Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
