

Personalized copy number and segmental duplication maps using next-generation sequencing
- PMID: 19718026
- PMCID: PMC2875196
- DOI: 10.1038/ng.437
Personalized copy number and segmental duplication maps using next-generation sequencing
Abstract
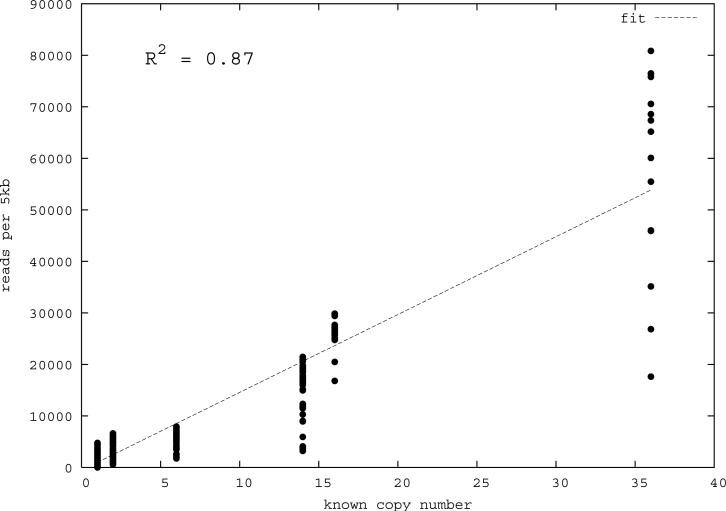
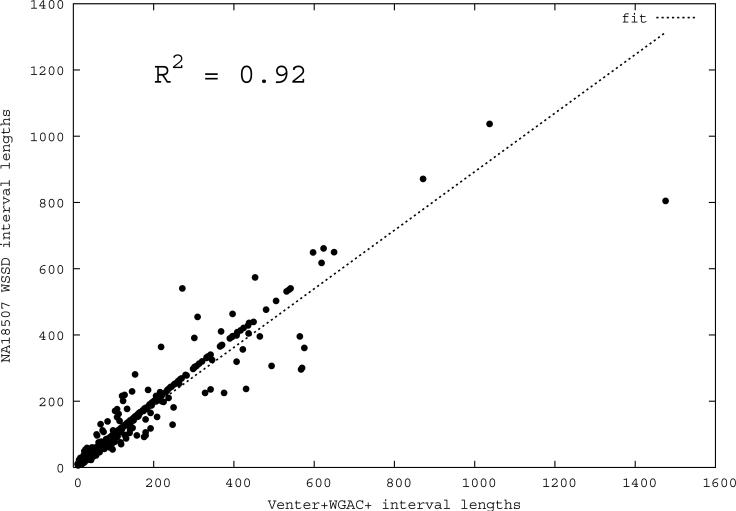
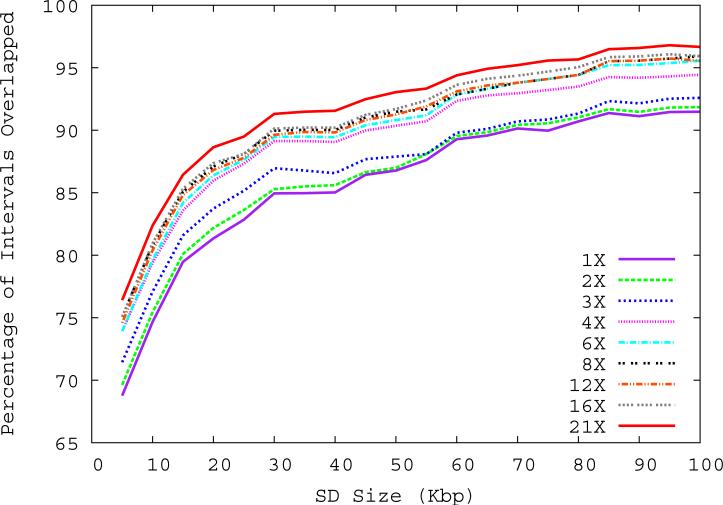
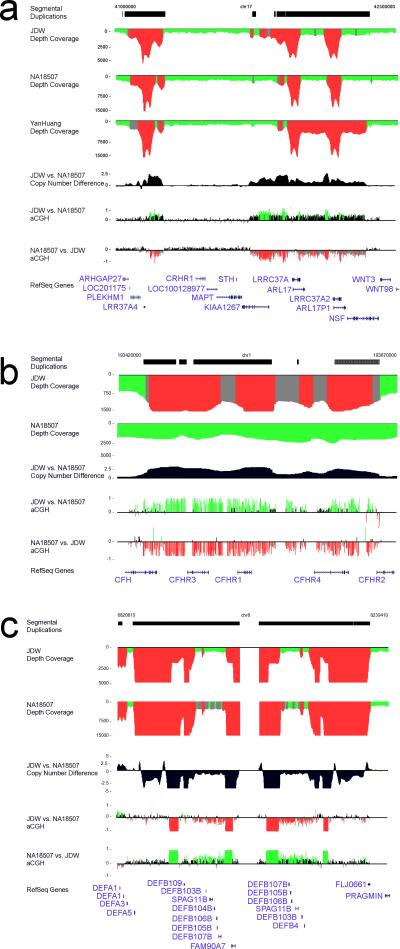
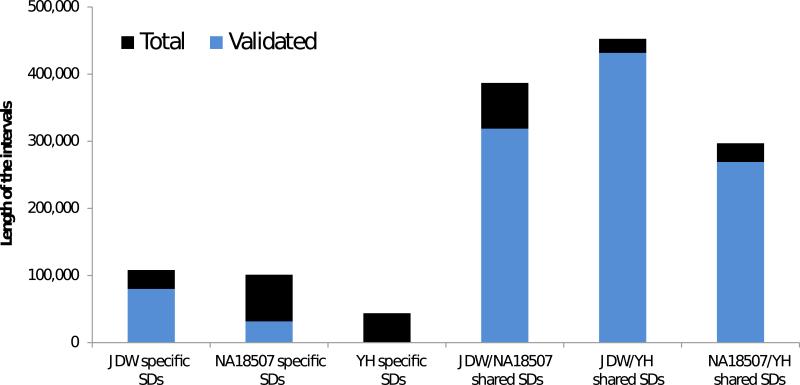
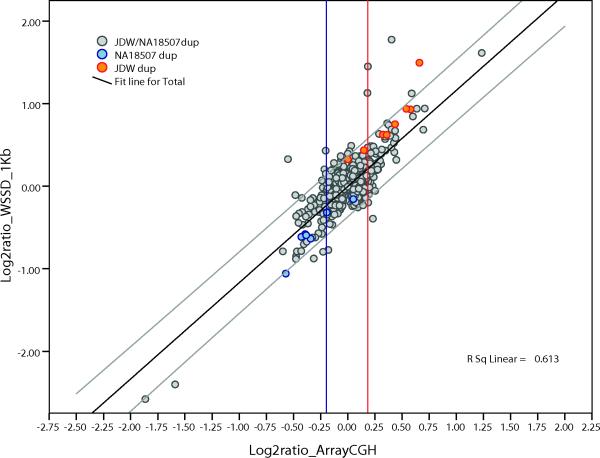
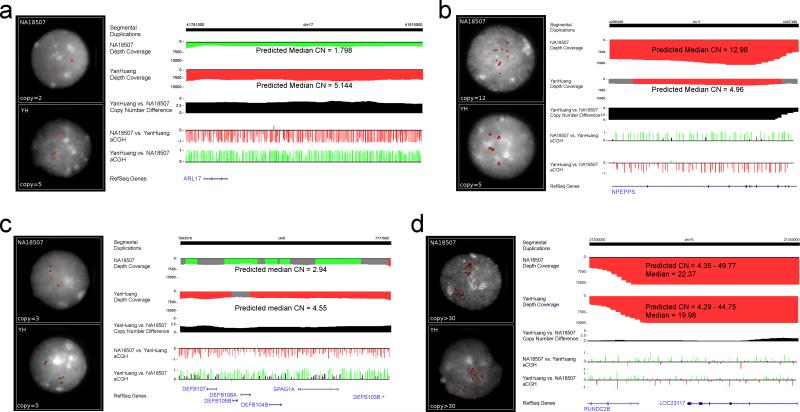
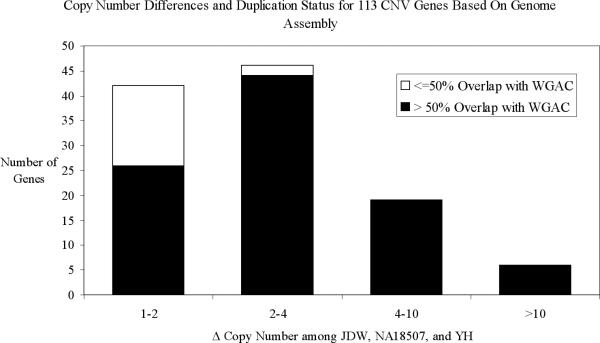
Despite their importance in gene innovation and phenotypic variation, duplicated regions have remained largely intractable owing to difficulties in accurately resolving their structure, copy number and sequence content. We present an algorithm (mrFAST) to comprehensively map next-generation sequence reads, which allows for the prediction of absolute copy-number variation of duplicated segments and genes. We examine three human genomes and experimentally validate genome-wide copy number differences. We estimate that, on average, 73-87 genes vary in copy number between any two individuals and find that these genic differences overwhelmingly correspond to segmental duplications (odds ratio = 135; P < 2.2 x 10(-16)). Our method can distinguish between different copies of highly identical genes, providing a more accurate assessment of gene content and insight into functional constraint without the limitations of array-based technology.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
