

Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy
- PMID: 19720722
- PMCID: PMC2768660
- DOI: 10.1093/brain/awp221
Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy
Abstract
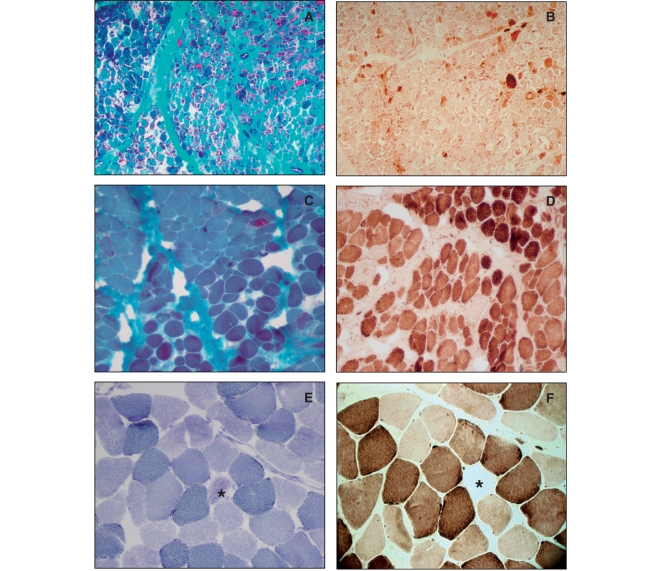
Childhood-onset mitochondrial encephalomyopathies are usually severe, relentlessly progressive conditions that have a fatal outcome. However, a puzzling infantile disorder, long known as 'benign cytochrome c oxidase deficiency myopathy' is an exception because it shows spontaneous recovery if infants survive the first months of life. Current investigations cannot distinguish those with a good prognosis from those with terminal disease, making it very difficult to decide when to continue intensive supportive care. Here we define the principal molecular basis of the disorder by identifying a maternally inherited, homoplasmic m.14674T>C mt-tRNA(Glu) mutation in 17 patients from 12 families. Our results provide functional evidence for the pathogenicity of the mutation and show that tissue-specific mechanisms downstream of tRNA(Glu) may explain the spontaneous recovery. This study provides the rationale for a simple genetic test to identify infants with mitochondrial myopathy and good prognosis.
Figures



References
-
- Bai RK, Perng CL, Hsu CH, Wong LJ. Quantitative PCR analysis of mitochondrial DNA content in patients with mitochondrial disease. Ann N Y Acad Sci. 2004;1011:304–309. - PubMed
-
- Chomyn A. In vivo labeling and analysis of human mitochondrial translation product. Methods Enzymol. 1996;264:197–211. - PubMed
-
- Debray FG, Lambert M, Chevalier I, Robitaille Y, Decarie JC, Shoubridge EA, et al. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics. 2007;119:722–733. - PubMed
-
- DiMauro S, Nicholson JF, Hays AP, Eastwood AB, Koenigsberger R, DeVivo DC. Benign infantile mitochondrial myopathy due to reversible cytochrome c oxidase deficiency. Trans Am Neurol Assoc. 1981;106:205–207. - PubMed
-
- DiMauro S, Nicholson JF, Hays AP, Eastwood AB, Papadimitriou A, Koenigsberger R, et al. Benign infantile mitochondrial myopathy due to reversible cytochrome c oxidase deficiency. Ann Neurol. 1983;14:226–234. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
