

Molecular pathophysiology of renal tubular acidosis
- PMID: 19721811
- PMCID: PMC2699831
- DOI: 10.2174/138920209787581262
Molecular pathophysiology of renal tubular acidosis
Abstract
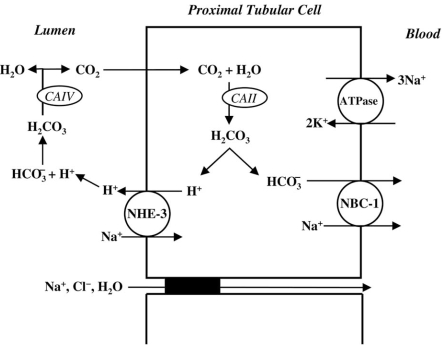
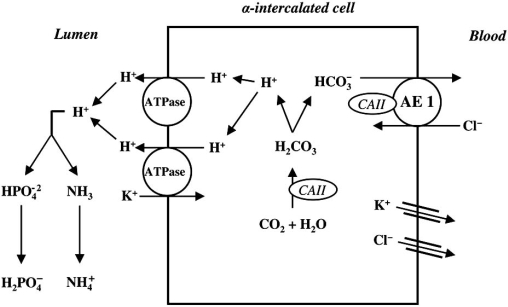
Renal tubular acidosis (RTA) is characterized by metabolic acidosis due to renal impaired acid excretion. Hyperchloremic acidosis with normal anion gap and normal or minimally affected glomerular filtration rate defines this disorder. RTA can also present with hypokalemia, medullary nephrocalcinosis and nephrolitiasis, as well as growth retardation and rickets in children, or short stature and osteomalacia in adults. In the past decade, remarkable progress has been made in our understanding of the molecular pathogenesis of RTA and the fundamental molecular physiology of renal tubular transport processes. This review summarizes hereditary diseases caused by mutations in genes encoding transporter or channel proteins operating along the renal tubule. Review of the molecular basis of hereditary tubulopathies reveals various loss-of-function or gain-of-function mutations in genes encoding cotransporter, exchanger, or channel proteins, which are located in the luminal, basolateral, or endosomal membranes of the tubular cell or in paracellular tight junctions. These gene mutations result in a variety of functional defects in transporter/channel proteins, including decreased activity, impaired gating, defective trafficking, impaired endocytosis and degradation, or defective assembly of channel subunits. Further molecular studies of inherited tubular transport disorders may shed more light on the molecular pathophysiology of these diseases and may significantly improve our understanding of the mechanisms underlying renal salt homeostasis, urinary mineral excretion, and blood pressure regulation in health and disease. The identification of the molecular defects in inherited tubulopathies may provide a basis for future design of targeted therapeutic interventions and, possibly, strategies for gene therapy of these complex disorders.
Keywords: Renal tubular acidosis; acid-base homeostasis; gene mutations.; molecular physiology; tubular transport.
Figures
References
-
- Herrin JT. Renal tubular acidosis. In: Avner ED, Harmon WE, Niaudet P, editors. Pediatric Nephrology. Philadelphia: Williams & Wilkins; 2003. pp. 757–776.
-
- Rodríguez-Soriano J. Renal tubular acidosis: the clinical entity. J. Am. Soc. Nephrol. 2002;13:2160–2170. - PubMed
-
- Smulders YM, Frissen PH, Slaats EH, Silberbusch J. Renal tubular acidosis. Pathophysiology and diagnosisArch. Arch. Intern. Med. 1996;156:1629–1636. - PubMed
-
- Gregory MJ, Schwartz GJ. Diagnosis and treatment of renal tubular disorders. Semin. Nephrol. 1998;18:317–329. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous