

Huntington's disease: the case for genetic modifiers
- PMID: 19725930
- PMCID: PMC2768966
- DOI: 10.1186/gm80
Huntington's disease: the case for genetic modifiers
Abstract
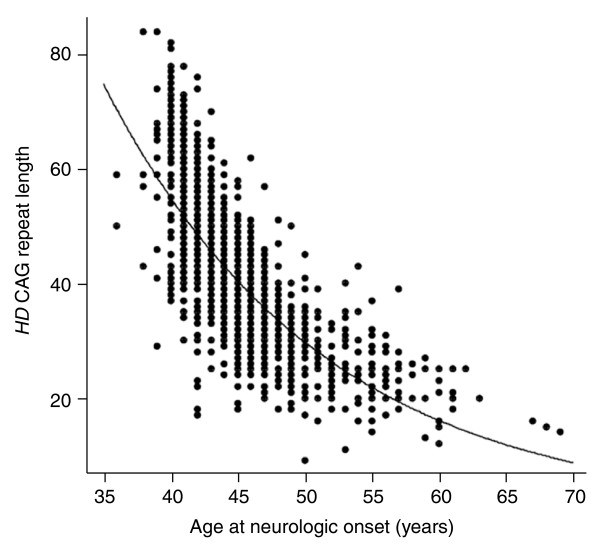
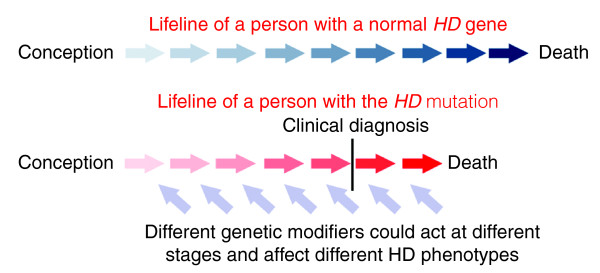
For almost three decades, Huntington's disease has been a prototype for the application of genetic strategies to human disease. HD, the Huntington's disease gene, was the first autosomal defect mapped using only DNA markers, a finding in 1983 that helped to spur similar studies in many other disorders and contributed to the concept of the human genome project. The search for the genetic defect itself pioneered many mapping and gene-finding technologies, and culminated in the identification of the HD gene, its mutation and its novel protein product in 1993. Since that time, extensive investigations into the pathogenic mechanism have utilized the knowledge of the disease gene and its defect but, with notable exceptions, have rarely relied for guidance on the genetic findings in human patients to interpret the relevance of findings in non-human model systems. However, the human patient still has much to teach us through a detailed analysis of genotype and phenotype. Such studies have implicated the existence of genetic modifiers - genes whose natural polymorphic variation contributes to altering the development of Huntington's disease symptoms. The search for these modifiers, much as the search for the HD gene did in the past, offers to open new entrées into the process of Huntington's disease pathogenesis by unlocking the biochemical changes that occur many years before diagnosis, and thereby providing validated target proteins and pathways for development of rational therapeutic interventions.
Figures
References
-
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. - DOI - PMC - PubMed
-
- Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Möller T, Tabrizi S. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205:1869–1877. doi: 10.1084/jem.20080178. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
